临床病例讨论——发热、皮下多发痛性结节
 |
 |
 |
 |
 |
病历摘要
患者 女,21岁。因结节性红斑、发热2年入院。
患者2年来无明显诱因反复出现持续发热、体温39℃左右,伴躯干部多发红色皮疹、皮下痛性结节(直径3~10 mm),无畏寒、寒战、咳嗽、腹痛等伴随症状。曾予多种抗生素治疗无效,于外院2次行皮下结节活检,病理提示“非化脓性脂膜炎”;予糖皮质激素(地塞米松最大剂量10 mg/d)及环磷酰胺(具体不详)治疗后体温恢复正常,皮下结节逐渐消退。但在激素减量过程中,病人病情多次反复,并出现双膝、双踝、双腕关节游走性疼痛,双上肢肌肉疼痛、乏力;病人无口腔溃疡、口眼干燥、光过敏及雷诺现象。既往体健,无类似家族史。
入院查体 T 36.6℃,BP 105/60 mmHg,皮肤、巩膜无黄染;腹部、臀部及四肢可见红色充血性皮疹及色素沉着斑;腹部及四肢可触及皮下硬结,直径10~20 mm,有触痛;无全身浅表淋巴结肿大;双肺呼吸音清,心界不大,心率80次/min、律齐;腹平软,肝脾肋下未及;关节无肿痛和畸形。
, http://www.100md.com
实验室检查 血常规:白细胞1.5×109/L,中性粒细胞58%,血红蛋白 114 g/L,血小板145×109/L;尿常规:蛋白(+);尿蛋白定量0.43 g/24 h;大便隐血(-)。肝肾功能:丙氨酸转氨酶(ALT)89 U/L,天冬氨酸转氨酶(AST)99 U/L,乳酸脱氢酶(LDH)787 U/L,总胆红素(TBIL)0.6 mg/dk,血清肌酐(Cr)0.6 mg/dk,尿素氮(BUN)13 mg/dk;凝血酶原时间(PT)12.6 s,激活的部分凝血活酶时间(APTT)26.5 s;血沉12 mm/1h,抗结核抗体(-);风疹病毒、巨细胞病毒、疱疹病毒、EB病毒抗体均阴性;C反应蛋白(CRP) 9.84 mg/L,血清铁蛋白1888
mg/dk,总补体(CH50)58.6
U/mk,抗核抗体(ANA)、抗双链DNA(dsDNA)、抗可提取核抗原(ENA)抗体、抗中性粒细胞胞浆抗体(ANCA)、抗心磷脂抗体(ACL)均阴性;甲胎蛋白、CA125、癌胚抗原、CA50、CA199、CA242基本正常;胸部、腹部及盆腔CT示:肝脾大,纵隔、腹膜后未见肿大淋巴结。上消化道造影(-),超声心动图正常,未见心包积液。
, 百拇医药
分析与讨论
本例为一年轻女性,慢性病程2年余,以反复发热、皮下结节为主要表现,予糖皮质激素治疗有效、但减量病情复发。仔细检查发现患者有系统性损害,包括肝酶谱升高及白细胞减少;但结缔组织病的相关抗体并无阳性提示。结合其为年轻患者,尤其是LDH升高明显,应首先考虑淋巴瘤的可能。需进一步行皮下结节、骨髓病理学检查。
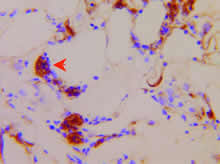
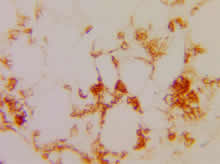
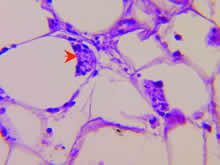
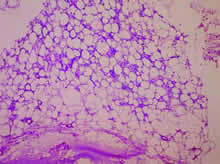
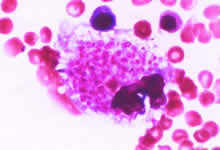
骨髓涂片示:组织细胞易见,并有吞噬血小板现象(图1);骨髓活检病理未见明显异常;下肢皮肤、皮下结节活检病理示:皮下脂肪小叶内有较多组织细胞、淋巴样细胞浸润,围绕在脂肪细胞周围(图2),组织细胞有明显吞噬现象、内含细胞碎片,呈典型“豆袋细胞”形态(图3),免疫组化CD45RO(++)(图4)、CD20(-)、CD68(+++)(图5)。
图1
骨髓涂片示:组织细胞易见,有吞噬血小板现象(May
, http://www.100md.com
& Giemsa stain ×1000)
图2皮肤活检示:脂肪小叶内较多组织细胞、淋巴样细胞浸润
(HE×40)
图3皮肤活检示:组织细胞有明显吞噬细胞碎片现象,呈“豆袋细胞”(↑)形态(HE×640)
图4
皮肤活检免疫组化示:CD45RO(++)提示T细胞来源
图5
皮肤活检免疫组化示:CD68(+++)提示组织细胞(↑)
诊断:组织细胞吞噬性脂膜炎(CHP)
, 百拇医药 依据皮下结节和骨髓病理学检查,诊断为CHP。那么,CHP是否能解释本例的临床全貌呢?复习文献,CHP最早由Winkekmann于1980年报告,指形态学良性的组织细胞浸润脂肪组织,并吞噬白细胞、红细胞和血小板的一类炎性疾病。本病可有多器官受累,表现为反复发热、浆膜炎、肝脾肿大等,与复发性结节性非化脓性脂膜炎(Weber-Christian综合征)的临床表现类似。Masayuki等在1999年通过对37例CHP临床资料的分析,总结了其临床病理特点。
1、病因学 最初认为CHP是一种病因不明的自发性脂膜炎。而近年多数学者认为CHP的发生与各种微生物感染有关,包括病毒、细菌、真菌乃至寄生虫感染。Akegre和Winkekmann发现CHP的皮下损害中有大量良性T淋巴细胞,故认为CHP可能是以T淋巴细胞增生为基础的反应性疾病或本身就是一种T淋巴细胞疾病。而Peters和Winkekmann报告了1例CHP患者最终进展为B细胞淋巴瘤,因而认为CHP是一种副肿瘤综合征或肿瘤的早期表现。Marzano则比较了4例CHP和3例皮下脂膜炎样T细胞淋巴瘤(SPTL)的免疫组化、分子生物学特点,同样认为CHP是进展为SPTL的前期阶段。
, 百拇医药
2、临床表现 CHP临床上以慢性、复发性可伴触痛的皮下结节为特点,主要分布于肢端,也可见于面、颈、躯干等处。全身表现包括反复高热,肝脾大(62.1%),淋巴结肿大,全血细胞减少,肝酶升高,凝血功能异常,乃至出现弥漫性血管内溶血(DIC),死亡率较高。少数病例可有黏膜溃疡及浆膜腔积液。病情进展迅速往往提示潜在恶性疾病的可能,尤其应首先与SPTL鉴别。
3、病理学 CHP的诊断及与恶性疾病的鉴别(尤其是SPTL)均依赖于病理。因此诊断上需要反复行皮下结节活检,并由经验丰富的病理科医师仔细检查。CHP受累脏器都可有组织细胞吞噬的特点,包括脂肪组织、骨髓、淋巴结及肝脾等。光镜下表现为小叶脂膜炎和灶性脂肪坏死,病灶内除分化良好的淋巴细胞浸润外,可见组织细胞吞噬红细胞、白细胞、血小板及核碎片等成分,形成特征性的“豆袋状”细胞。免疫组织化学检查显示浸润细胞来源为T淋巴细胞,且组织细胞的标记抗原CD68阳性。
本例的临床特点与文献报道基本相符,且病程已2年余,因而恶性肿瘤的可能性不大,病理学检查未见异形淋巴细胞,亦不支持SPTL的诊断。同时本例自身免疫病相关抗体指标阴性,筛除了该病的可能,也除外了继发于结缔组织病的脂膜炎的可能。
, http://www.100md.com
4、治疗和预后 改善CHP预后的治疗策略仍是临床上的难题。对发病机制的研究显示,组织细胞的吞噬功能是由增生的T淋巴细胞释放的细胞因子所激活,所以针对淋巴瘤采用积极联合化疗方案是必要的,以达到有效抑制T细胞增生的目的。Masayuki总结了37例CHP病死率为43.2%(16/37例),但1989年前CHP病死率(12/20例,60.0%)明显高于1990年后的病死率(4/17例,23.5%)。而如此的差异与不同的治疗方案直接相关。在1989年之前,CHP的治疗方案为单独应用糖皮质激素或与环磷酰胺合用;而在1990年之后,采用CHOP方案(环磷酰胺、柔红霉素、长春新碱、泼尼松龙),或包括环孢素A的联合化疗方案逐步成为CHP的治疗首选。6例采用CHOP方案治疗的CHP患者仅有1例死亡,而采用环孢素A联合化疗的8例患者亦仅有1例死亡。这些结果充分证实了CHP的治疗应针对T淋巴细胞增加化疗强度。所以我们决定对这例病人采用CVP化疗方案。
患者入院后,接受泼尼松40 mg/d及非类固醇类抗炎药(NSAID)对症退热治疗后,体温基本正常,但皮下结节未消退。遂给予CVP方案(环磷酰胺600 mg d1,长春新碱2 mg d1,泼尼松40 mg/d)化疗,患者体温正常、皮下结节逐渐消退。目前对该患者定期随访,继续联合化疗,患者病情一直稳定。
点评
本文报告了1例临床少见的CHP。虽然CHP是Weber-Christian综合征的一个亚型,但有其独特的临床表现,包括发热、皮下结节、肝损害及粒细胞减少等。进一步行皮肤、骨髓病理检查对CHP的诊断意义重大。CHP的治疗方案提倡用联合化疗,同时需要严密随访,警惕其发展为恶性疾病,特别是SPTL的可能。
刘晓红, 百拇医药(李梦涛 张奉春 唐福林(风湿免疫科)渠涛(皮肤科))