关键词:夏科-马里-图思病;;基因;点突变;聚合酶链反应-多态现象;单链构象
【摘要】 目的 研究我国夏科-马里-图思病(CMT)Cx32、MPZ 和PMP22基因点突变的特点。方法 应用聚合酶链反应-单链构象多态性(PCR-SSCP)结合DNA序列分析检测30个CMT家系的患者及50名非血缘关系的正常对照的Cx32、MPZ和PMP22基因的编码外显子。结果 4个家系(3个为X-连锁隐性遗传)有4种不同的Cx32基因点突变(13.3%),其中1个家系为188位的苏氨酸被丙氨酸置换是未见报道的新突变。 1个家系(3.3%)有MPZ基因点突变,未发现PMP22基因的点突变。结论 Cx32、MPZ和PMP22基因的点突变占CMT致病原因的16.7%。我国也存在不少X-连锁遗传的家系(>10.0%),对可疑的家系(家系中患者无男传男的现象)应首先进行Cx32 基因的点突变检测。我国的CMT发病率低可能与其基因突变特点有关。
Point mutation of Cx32, MPZ and PMP22 genes in Chinese Charcot-Marie-Tooth disease
XIAO Jianfeng* , TANG Beisha, XIA Jiahui, et al. * Department of Neurology, Xiang Ya Hospital, Hunan Medical University, Changsha 410008
【Abstract】 Objective To study thecharacteristics of the point mutations of Cx32, MPZ and PMP22 genes in Chinese Charcot-Marie-Tooth disease(CMT). Methods The coding exons of Cx32, MPZand PMP22 genes were screened in 30 families of Chinese CMT and 50 unrelated normalpersons by polymerase chain reaction-single strand conformation polymorphism. Results Four(13.3%) different point mutations (3 had been described previously) were found in 4families (3 with X-linkage ressesive transmission). One of the point mutations, Thr188Ala,which was detected in the only X-linkage dominant transmission family, was a novelmutation. One mutation (3.3%) in the exon 3 of MPZ gene was identified in a CMT1 family.There were no PMP22 gene mutations in these patients. Conclusions About16.7% CMT had point mutations in the Cx32, MPZ and PMP22 genes, and the mutations mainlybelonged to Cx32 gene mutations. Mutational screening had to start with Cx32 gene in thepossible X-linkage family (especially if no male-to-male transmission was observed).Thelow prevalence of Chinese CMT might be due to the different point mutations in thesegenes.
【Key words】 Charcot-Marie-Tooth disease Genes Pointmutation PCR-SSCP
夏科-马里-图思病(Charcot-Marie-Tooth disease, CMT)是一类常见的以胫前腓骨肌萎缩为主要表现的周围神经系统单基因遗传病,欧美国家发病率高达1/2500[1] ,遗传方式和临床表现复杂多样,主要为常染色体显性遗传(AD),临床依据电生理及病理改变分为脱髓鞘型(CMT1)和轴索型(CMT2)。目前研究发现,CMT的基因分型至少有13型,其中4型已明确了致病基因,均为与髓鞘蛋白有关的基因[2] ,它们是周围神经髓鞘蛋白(PMP)22基因(定位于17p11.2)、髓鞘蛋白零基因(MPZ,1q22)、间隙连接蛋白(Connexin)32基因(Xq13.1)、早期生长反应蛋白(EGR)2基因(10q21.1~22.1)。国外研究发现,该四型基因的点突变约占CMT致病原因的10%[3] ,我国至今虽已有200多个CMT家系的报道,但发病率相对较低,原因未明。为此,我们检测了30个CMT家系的PMP22、MPZ、Cx32基因的点突变的情况,旨在明确我国CMT该三型基因点突变的分布状况,并探讨基因突变型与临床表现型的关系。 资料和方法
30个家系为我科及中国医学遗传学国家重点实验室的中国遗传病资源保藏中心所收集的病例,来自全国8个省,诊断标准为:缓慢进展的双下肢腓肠肌为主的对称性无力及萎缩,伴腱反射减退和(或)末梢型感觉减退及弓形足,如正中神经传导速度<38m/s或腓肠神经活检示脱髓鞘者诊断为CMT1型;正中神经传导速度≥38m/s或腓肠神经活检示轴索变性为主者诊断为CMT2型。功能评分参见文献[4],分0~8级。另取无血缘关系的正常成人50名作为对照。
二、方法
用聚合酶链反应-单链构象多态性(PCR-SSCP)结合DNA序列分析检测各基因的编码外显子。采血10 ml,按常规酚/氯仿法抽提DNA,每个家系取1例患者作检测,MPZ、PMP22基因的引物序列见文献[5]和[6],Cx32基因的引物序列见表1,PCR总反应体积为 20 μl,其中dNTPs的终浓度为200 nmol/L,引物的终浓度为1 μmol/L,Mg2+ 的终浓度为1.5 mmol/L, gDNA为100ng, Taq酶为0.8 U,反应在PE9600 PCR仪进行,条件为: 95℃预变性2分30秒,然后95℃变性15秒,退火30秒,72℃延伸45秒,28个循环后于72℃再延伸5分钟,取5 μl经聚丙烯酰胺凝胶电泳检测产物,如特异性好,则加15μl变性载样液,混合后96℃变性5分钟,立即置冰上5分钟,取10 μl左右上样于含5%甘油的6%~10%的聚丙烯酰胺凝胶中,250V、 4℃电泳16~24小时,银染观察结果,有异常泳带者取家系全部成员重复2次,并同时检测50名正常对照,排除多态后,手工抽提纯化模板,直接法在377型测序仪(美国PE公司产)进行DNA序列分析2次。测序结果在基因突变数据库中进行比较分析。
表1 Cx32基因PCR-SSCP引物序列表
| 引物名 | 序列(5′→3′) | 扩增片段 (bp) | 退火温度 |
| C1F | GGTGTTTGCAGGTGTGAATG | 230 | 60℃ |
| C1R | ATTGGTCATAGCAAACGCTG | ||
| C2F | CCTTCATCTGCAACACACTC | 273 | 60℃ |
| C2R | ACACCACGCTGATGACATAG | ||
| C3F | GAGGTGAAGAGGCACAAGG | 215 | 60℃ |
| C3R | AAGACGGTTTTCTCGGTGG | ||
| C4F | GGTCAAGTGCGACGTCTAC | 264 | 60℃ |
| C4R | CTCAGCAGCTTGTTGATCTC | ||
| C5F | CCGCCTCTCACCTGAATAC | 247 | 60℃ |
| C5R | AGTAATCCCCAGCAGGCAG |
结果
30个家系存活患者共70例,男性53例,女性17例,男女之比为3∶1。CMT1型20个家系57例,CMT2型5个家系5例,无法分型者 5个家系8例。AD遗传9个家系30例,常染色体隐性遗传(AR)4个家系8例,X-连锁遗传3个家系14例,不能确定者4个家系8例,散发10个家系10例。家族史阳性率为66.6%。
二、 SSCP检测和基因突变情况
1. Cx32、MPZ、PMP22基因各发现5、3、2个家系有异常泳带,排除多态后仍有异常的家系分别有4、1、0个。
2. 5个有基因突变的CMT家系的先证者基因突变情况见表2。
表2 CMT先证者基因突变一览表
| 基因 | 家系编号 | 诊断 | 评分 | 遗传方式 | 突变位点 | 氨基酸改变 | 酶切改变 | 报道者 |
| Cx32 | CMT004 | CMT1 | 3 | XR* | 15CGG→CAG | Arg→Gln | 增加MspI | 文献[7] |
| CMT012 | CMT1 | 4 | XD | 188ACC→GCC | Thr→Ala | 增加BctI | 本研究作者 | |
| CMT018 | CMT1 | 3 | 散发* | 164CGG→CAG | Arg→Gln | 增加MspI | 文献[8] | |
| CMT030 | CMT1 | 3 | XR* | 183CGC→CAC | Arg→His | 减少AciI | 文献[8] | |
| MPZ | CMT007 | CMT1 | 4 | AD | 124ACG→ATG | Thr→Met | 减少NspI | 文献[9] |
注:*为本研究与国外报道的家系遗传方式有所不同
3. 12号家系DNA序列图见图1。
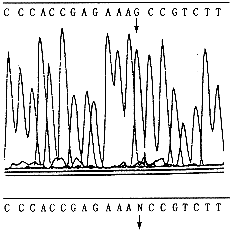


箭头指示突变位点。上图:半合子的先证者;中图:先证者的母亲(为杂合子患者),N表示A562 →G;下图:先证者的正常儿子
图1 12号家系3名成员DNA序列图
CMT是一大类临床表现和遗传致病机制均复杂的疾病,尽管CMT基因分型至少有13型,但CMT1型的AD遗传的70%和散发病例的90%是由于包含PMP22基因在内的1.5 Mb的大片段重复突变所致,部分是由于MPZ、PMP22基因的点突变所致。而X-连锁遗传的大部分CMT患者为Cx32基因的点突变所致,通过PMP22基因的重复突变检测和该三型基因的点突变检测可对50%~60%的CMT患者作出基因诊断[2] 。
虽然基因点突变的检测方法很多,但简单易行的方法仍属SSCP,我们共设计了15对引物,结果显示检测效果好。我们研究发现:Cx32基因的点突变发生率较多,占CMT总数的 13.3%(4/30),3.3%(1/30)的患者是MPZ基因突变,未发现PMP22的点突变,而国外资料显示,Cx32、MPZ、PMP22基因的点突变分别占5.4%、0.7%、2.7%[3] ,这种差异一方面可能是家系选择上的偏差,另一方面可能是我国CMT的发病机制在分子水平上与国外确实存在一定的差异,这可能是我国CMT的发病率较低的原因之一。同时也提示,我国的CMT患者如存在X-连锁遗传的可能(家系中的患者无男传男的遗传方式)应首先行Cx32基因的突变检测。
Cx32基因属间隙连接蛋白基因家族的β1类,故又名GJB1。其cDNA全长1574 bp,仅有1个编码外显子,编码含 283个氨基酸、分子量约为32 000的蛋白间隙连接32。该蛋白有4个跨膜区,在髓鞘的郎飞结和Schmidt-Lanterman切迹有正常表达。其功能可能在施万细胞胞浆的皱褶间形成细胞内间隙连接。目前在CMT患者中已检测出Cx32基因的 73种不同的突变位点和形式。我们发现的 4种Cx32的突变形式中有3种国外已有报道[7,8] ,12号家系的562位碱基G被A置换,导致Cx32蛋白的 188位氨基酸发生错意突变,即苏氨酸被丙氨酸替代,是一种新的突变形式,该突变虽然氨基酸的性质改变不大,但由于发生在Cx32蛋白的第二个膜外区,该位点可能非常关键,从而导致了较重的临床表现及典型的X-连锁显性遗传(女性杂合子的症状也较重),与其不同的是,同一功能域的18号和29号家系发生的错意突变,以及 3号家系的膜内区的错意突变,临床却均表现为男性发病,无女性患者,突变检测发现女性杂合子为表型正常的携带者(符合X-连锁隐性遗传),与国外报道的这些位点的突变病例不尽相同;且所有CMTX1基因型的患者均表现为CMT1型,而非国外学者所报道的以CMT2型为主[9] ,也表明种族间的差异较明显。我国的X-连锁遗传的CMT以隐性遗传为主,这可能是我国CMT男性患者更多的原因,也可能是我国CMT的发病率较低的又一原因,其意义值得探讨。其中2个突变(第164及183位氨基酸)是西班牙的CMT家系的突变热点[8] ,两民族有无历史渊源有待进一步研究。而3个突变均发生在精氨酸上的意义也有待阐明。
MPZ基因的突变虽只有1例,但其位于 3号外显子,而国外研究发现,该外显子正是突变的热点[2] ,因此,该外显子也应作为我国CMT突变检测的重点。我们未在该组病例中检测到PMP22基因的点突变,间接表明我国CMT的PMP22基因突变可能主要是大片段的重复突变。不同基因的不同位点及不同形式的突变的临床意义有待更深入研究。
本课题由国家 863计划项目资助(No.z19-02-02-02)
参考文献
1 Murakami T, Garcia CA, Reiter LT, et al. Charcot-Marie-Toothdisease and related inherited neuropathies. Medicine (Baltimore), 1996, 75: 233-250.
2 萧剑锋. 腓骨肌萎缩症的分子遗传学研究进展.国外医学神经病学神经外科学分册,1998, 25: 182-185.
3 Janssen EAM, Kemp S, Hensels GW, et al. Connexin 32 gene mutations in X-linkeddominant Charcot-Marie-Tooth disease (CMTX1). Hum Genet, 1997,99:501-505.
4 Birouk N, Leguern E, Maisonobe T, et al. X-linked Charcot-Marie-Tooth disease withconnexin 32 mutations: clinical and electrophysiologic study. Neurology,1998,50:1074-1082.
5 Nelis E, Timmerman V, Jonghe PD, et al. Rapid screening of myelin genes in CMT1patients by SSCP analysis: identification of new mutations and polymorphisms in the P0gene. Hum Genet, 1994,94:653-657.
6 Roa BB, Garcia CA, Suter U, et al. Charcot-Marie-Tooth disease type 1A. Associationwith a spontaneous point mutation in the PMP22 gene. N Engl J Med,1993,329:96-101.
7 Fairweather N, Bell C, Cochrane S, et al. Mutations in the connexin 32 gene inX-linked dominant Charcot-Marie-Tooth disease. Hum Mol Genet, 1994, 3:29-34.
8 Bort S, Nelis E, Timmerman V,et al. Mutational analysis of the MPZ, PMP22 and Cx32genes in patients of Spanish ancestry with Charcot-Marie-Tooth disease and hereditaryneuropathy with liability to pressure palsies. Hum Genet, 1997,99:746-754.
9 Silander K, Meretoja P, Pihko H, et al. Screening for connexin 32 mutations in Charcot-Marie-Tooth disease families with possible X-linked inheritance. HumGenet,1997,100:391-397.
(收稿:1998-10-06 修回:1999-03-24) , 百拇医药