H+/K+-ATPΟΗ“÷÷ΤΦΝΒΡ―–ΨΩΫχ’Ι
Ής’ΏΘΚΒ‘Ϋ®Ιζ ψΤΕ§ ≥ΧΟ°…ζ
ΒΞΈΜΘΚ…ρ―τ“©ΩΤ¥σ―ß“©ΈοΚœ≥… “Θ§…ρ―τ 110015
ΙΊΦϋ¥ ΘΚH+/K+-ATPΟΗ“÷÷ΤΦΝΘΜ“÷Υα“©ΈοΘΜΩΙάΘ―ώΘΜΩ…Ρφ–ΆΘΜ≤ΜΩ…Ρφ–ΆΘΜΉς”ΟΜζάμ
÷–Ιζ“©ΈοΜ·―ß‘”÷Ψ000222 ’Σ“Σ Ϋι…ήΝΥ“÷Υα“©ΈοH+/K+-ATPΟΗ“÷÷ΤΦΝΒΡ―–ΨΩ«ιΩωΚΆΖΔ’ΙΖΫœρΘ§≤ΔΕ‘“―Ψ≠±®ΒάΒΡH+/K+-ATPΟΗ“÷÷ΤΦΝΒΡΉς”ΟΜζάμΫχ––ΝΥ±»Ϋœ.
Advance on the Research and Development of H+/K+-ATPase Inhibitors
Zhai Jianguo,Yan Dong,Cheng Maosheng
, http://www.100md.com
(Laboratory of Drug Synthesis,Shenyang Pharmaceutical University,Shenyang 110015)
Abstract This is about advance on the research and development of H+/K+-ATPase inhibitors.The mechanism of action of H+/K+-ATPase inhibitors was also discussed.
Key words H+/K+TPase inhibitors;antisecretory;antiulcer agents;reversible;irreversible;mechanism
, ΑΌΡ¥“Ϋ“©
H+/K+-ATPΟΗ“÷÷ΤΦΝΘ§Φ¥÷ Ή”±Ο“÷÷ΤΦΝ(proton pump inhibitors,PPI), «“Μάύ–¬–ΆΒΡ“÷÷ΤΈΗΥαΖ÷ΟΎΒΡ“©Έο.H+/K+-ATPΟΗ(÷ Ή”±Ο)Θ§ΈΜ”Ύ±ΎœΗΑϊΒΡΙήΉ¥Ρ“≈ίΚΆΖ÷ΟΎΙήΡΛ…œΘ§≤Έ”κΈΗΥαΖ÷ΟΎΒΡΉνΚσ“ΜΗωΜΖΫΎΘ§“÷÷Τ¥ΥΟΗΒΡΜν–‘Θ§Ω…“÷÷ΤΗς÷÷“ρΥΊ“ΐΤπΒΡΈΗΥαΖ÷ΟΎ.PPI÷ς“Σ”Ο”Ύ÷ΈΝΤΈΗάΘ―ώΓΔ °Εΰ÷Η≥ΠάΘ―ώΓΔΖ¥Νς–‘ ≥Ιή―ΉΚΆΉτ-ΌΙ œΉέΚœ÷ΔΒ»”κΈΗΥαΖ÷ΟΎ ßΒς”–ΙΊΒΡΦ≤≤Γ.’βάύ“©ΈοΫœΉιΑΖH2- ήΧεόΉΩΙΦΝΦΑΤδΥϋ“÷÷ΤΈΗΥαΖ÷ΟΎΒΡ“©ΈοΘ§ΨΏ”–Οςœ‘ΒΡ”≈‘Ϋ–‘Θ§»γ―Γ‘ώ–‘ΗΏΓΔΝΤ–ßΚΟΓΔΗ±Ής”Ο…ΌΓΔ”κΩΙ…ζΥΊ≈δΈιΒΡΗ¥ΖΫ÷ΤΦΝΩ…œϊ≥ΐ”ΡΟ≈¬ίΗΥΨζ(ΈΗάΘ―ώ÷¬≤ΓΨζ)Β».PPI“―”–4Ηω–¬“©≈ζΉΦ…œ –Θ§’ΐ‘ΎΜρΦ¥ΫΪΫχ––ΝΌ¥≤ ‘―ιΒΡ“≤”–≤Μ…Ό.ΗυΨίΤδ”κH+/K+-ATPΟΗΉς”ΟΒΡ≤ΜΆ§ΖΫ ΫΘ§÷ς“ΣΖ÷ΈΣΩ…Ρφ–ΆΚΆ≤ΜΩ…Ρφ–ΆΝΫάύ.ΡΩ«ΑPPIΒΡ―–ΨΩΕύΈΣ―Α’“Ω…Ρφ–ΆΜν–‘“©Έο.
1 ≤ΜΩ…Ρφ–ΆPPI
, http://www.100md.com
≤ΜΩ…Ρφ–ΆPPI «“Μ÷÷«Α“©Θ§Υϋ‘ΎΈΗ±ΎœΗΑϊΒΡΥα–‘ΜΖΨ≥÷–ΉΣΜ·ΈΣ¥ΈΜ«ΥαΜν–‘ΧεΘ§”κH+/K+-ATPΟΗ“‘ΕΰΝρΦϋΫαΚœΘ§–Έ≥…ΟΗ-“÷÷ΤΦΝΗ¥ΚœΈοΘ§≤ΜΩ…ΡφΒΊ“÷÷ΤH+/K+-ATPΟΗΒΡΜν–‘Θ§ΫΒΒΆΈΗΥαΖ÷ΟΎ.’ΐ‘Ύ―–ΨΩΒΡ≤ΜΩ…Ρφ–ΆPPI÷ς“Σ”–±Ϋ≤ΔΏδΏράύΓΔ≥μ‘”≤ΔΏδΏράύΓΔ»Γ¥ζΏδΏράύΚΆΡαΙ≈ΕΓθΘΑΖάύΒ»―ή…ζΈο.
1.1 ±Ϋ≤ΔΏδΏράύ
¥ΥάύPPIΒΡΜν–‘œ»ΒΦΜ·ΚœΈοtimeprazole(1a)Θ§ «»πΒδAstra Hassle―–ΨΩΥυ”Ύ1978Ρξ‘Ύ“÷Υα“©ΈοΒΡ…Η―Γ÷–ΖΔœ÷ΒΡΘ§“ρΒ± ±ForteΒ»»Υ“―ΖΔœ÷ΝΥH+/K+-ATPΟΗΓ≤1Γ≥Θ§‘χΆΤ≤β(1a)ΒΡ“÷ΥαΜζάμΈΣ“÷÷ΤH+/K+-ATPΟΗΒΡΜν–‘.÷°Κσ¥Υ―–ΨΩΥυΚœ≥…ΝΥpicoprazole(1b)ΚΆomeprazole(1c)Γ≤2,3Γ≥Θ§≤Δ≤ϊΟςΤδΕ‘H+/K+-ATPΟΗΒΡΉς”ΟΜζ÷Τ.(1c)”Ύ1988Ρξ≈ζΉΦ…œ –“‘ΚσΘ§“Μ÷±ΈΜ”Ύ άΫγ≥©œζ“©ΈοΒΡ«ΑΝ–Θ§ΤδΒΞ“ΜΒΡΉσ–ΐΙβ―ß“λΙΙΧεperprazole“―Ϋχ»κΔσΤΎΝΌ¥≤ ‘―ιΚσΤΎΘ§≤Δ‘ΎΟάΙζΜώΒΟ÷ΤΦΝΉ®άϊ.»’±ΨTakedaΙΪΥΨΒΡlansoprazole(1d)Θ§Β¬ΙζBykGuldenΙΪΥΨΒΡpantoprazole(1e)ΡΤ―ΈΚΆ»’±ΨEisaiΙΪΥΨΒΡrabeprazole(E3810,1f)ΡΤ―ΈΘ§Ζ÷±π”Ύ1991Θ§1994ΚΆ1997Ρξ≈ζΉΦ…œ –.ΚΪΙζΔρ-Yang“Ϋ“©ΙΪΥΨΒΡIY-81149(1g)Γ≤4Γ≥Θ§“÷÷Τ”ΡΟ≈¬ίΗΥΨζΒΡΉς”Ο«Ω”Ύ(1c)Θ§ΑκΥΞΤΎΕΧΘ§Ε‘―Σ“ΚΈΗΟΎΥΊΒΡ≈®Ε»”ΑœλΫœ–ΓΘ§ΩΙΈΗάΘ―ώΉς”Ο «(1c)ΒΡ3±ΕΘ§«“≤Μ≤ζ…ζΩΙ“©–‘Θ§ΈόΟςœ‘Η±Ής”ΟΘ§Α≤»Ϊ–‘ΫœΚΟΘ§’ΐΫχ––ΔρΤΎΝΌ¥≤ ‘―ι.ΨΏ”–(1a)Μυ±ΨΫαΙΙΒΡPPIΜΙ”–ΚήΕύ’ΐ‘ΎΩΣΖΔΙΐ≥Χ÷–Θ§»γdisuprazole(1h),SK&F-95601(1i)ΚΆRo-18-5364(1j)Β».
, ΑΌΡ¥“Ϋ“©
Ε‘œ»ΒΦΜ·ΚœΈο(1a)ΒΡΫαΙΙ–ό Έ”–“ΜΕ®ΒΡΨ÷œό–‘Θ§–μΕύΙΪΥΨΕ‘±Ϋ≤ΔΏδΏρΜΖΚΆΏΝύΛΜΖΒΡΩ…Χφ¥ζ–‘Ϋχ––ΝΥ―–ΨΩΘ§ΖΔœ÷ΝΥ–μΕύ“÷ΥαΚΆΩΙάΘ―ώΉς”ΟΕΦΚήΚΟΒΡPPI.»γ»’±ΨToaEiyoΙΪΥΨΒΡTY-11345(2)Γ≤5Γ≥Θ§“÷ΥαΉς”Ο «(1c)ΒΡ2ΓΪ3±ΕΘ§ΩΙάΘ―ώΉς”Ο «(1c)ΒΡ3ΓΪ15±ΕΘ§’ΐΫχ––ΔρΤΎΝΌ¥≤ ‘―ι.ύΉύΛΦΉΥαθΞ―ή…ζΈο(3)Γ≤6Γ≥ΩΎΖΰΚΆΨ≤¬ωΉΔ…δΨυ”–ΈΗ’≥ΡΛœΗΑϊ±ΘΜΛΉς”ΟΘ§ΤδΩΙάΘ―ώΉς”Ο «ΉιΑΖH2- ήΧεόΉΩΙΦΝΈςΟΉΧφΕΓΒΡ3±Ε.±ΫΑΖ―ή…ζΈοNC-1300(4a)ΚΆNC-1300-O-3(leminoprazole,4b)Γ≤7Γ≥Θ§Φφ”–“÷÷ΤΈΗΥαΖ÷ΟΎΚΆΈΗ’≥ΡΛœΗΑϊ±ΘΜΛΉς”Ο.(4b)”…»’±ΨNippon ChemipharΙΪΥΨ―–÷ΤΘ§ΥϋΒΡΩΙάΘ―ώΜν–‘”κ(1c)œύΥΤΘ§Ε‘H+/K+-ATPΟΗΒΡ“÷÷Τ≥÷–χ ±ΦδΫœ≥ΛΘ§”–ΩΙ”ΡΟ≈¬ίΗΥΨζΉς”ΟΘ§Ε‘÷– ύΚΆΉ‘÷ς…ώΨ≠œΒΆ≥ΓΔ–Ρ―ΣΙήœΒΆ≥Β»ΈόΟςœ‘”ΑœλΘ§’ΐΫχ––ΔσΤΎΝΌ¥≤ ‘―ι.“―±®ΒάΒΡΜΙ”–S-3337(4c)Β».1,2,3,4-ΥΡ«βύ≠Ώχ―ή…ζΈοOPC-22381(5a)ΚΆOPC-22575(5b)Γ≤8Γ≥Β»Θ§“÷ΥαΉς”ΟΫœ»θ.
, http://www.100md.com
±Ϋ≤ΔΏδΏράύPPIΒΡ¥ζ±μΜ·ΚœΈο «(1c)Θ§ΥϋΫχ»κΈΗΒΡ±ΎœΗΑϊΚσΘ§‘ΎΖ÷ΟΎΙή÷–H+ΒΡΉς”Οœ¬Θ§ΖΔ…ζΖ÷Ή”ΡΎ«ΉΚΥΖ¥”ΠΘ§–Έ≥…¬ί÷–ΦδΧεΘ§ΉΣΜ·≥…¥ΈΜ«ΥαΓΔ¥ΈΜ«θΘΑΖΜν–‘Μ·ΚœΈοΘ§”κH+/K+-ATPΟΗΠΝ―«Μυ÷–ΒΡCys813ΚΆCys892≤–Μυ“‘ΕΰΝρΦϋΫαΚœΘ§–Έ≥…ΟΗ-“÷÷ΤΦΝΗ¥ΚœΈοΘ§“÷÷ΤH+/K+-ATPΟΗΒΡΜν–‘.ΤδΥϋΒΡ±Ϋ≤ΔΏδΏράύPPIΕ‘H+/K+-ATPΟΗΒΡΉς”ΟΜζ÷ΤάύΥΤ”Ύ(1c)Θ§“≤ «”κΡ≥Cys≤–ΜυΫαΚœ.
±Ϋ≤ΔΏδΏράύPPIΕ‘H+/K+-ATPΟΗΒΡ“÷÷ΤΉς”ΟΫœ«ΩΘ§Εχ«“≥÷ΨΟΘ§Ε‘ΈΗΥα“÷÷ΤΒΡ≥ΧΕ»Ϋœ…νΘ§“ΜΑψΕΧΤΎ”Ο“©ΨΆΡή÷Έ”ζΈΗάΘ―ώΜρ °Εΰ÷Η≥ΠάΘ―ώΘΜΒΪ≥ΛΤΎ”Ο“©Θ§“Ή“ΐΤπ”κΈΗΥα»±ΖΠ”–ΙΊΒΡΗ±Ζ¥”ΠΘ§»γœϊΜ·≤ΜΝΦΓΔΗΏΈΗΟΎΥΊ―Σ÷ΔΓΔ≥Π »ΗθœΗΑϊ‘ω…ζΒ».≤ΩΖ÷≤Γ»Υ”Ο“©Κσ≥ωœ÷Εώ–ΡΓΔ≈ΜΆ¬ΓΔ’ΆΤχΓΔΗΙ–ΚΒ»≤ΜΝΦΖ¥”ΠΘ§“ΜΑψ≤Μ–ηΦθΝΩΜρΆΘ÷Ι÷ΈΝΤ.≥ΐ(1e)ΆβΘ§±Ϋ≤ΔΏδΏράύPPIΜΙΡή“÷÷ΤΗΈΈΔΝΘΧεœΗΑϊ…ΪΥΊP450ΟΗœΒΒΡ“©Έο¥ζ–ΜΉς”ΟΘ§ΫΒΒΆœψΕΙΥΊάύΩΙΡΐ“©ΓΔΑ≤Ε®ΓΔ±ΫΆΉ“ρΒ»“©ΈοΒΡ«ε≥ΐ¬ .
, http://www.100md.com
1.2 ≥μ‘”≤ΔΏδΏράύ
»’±ΨTokyo TanabeΙΪΥΨΒΡTU-199(6)Γ≤9Γ≥“÷ΥαΚΆΩΙάΘ―ώΉς”ΟΕΦ≥§Ιΐ(1c)Θ§«“Μ·―ßΈ»Ε®–‘ΚΟΘ§’ΐΫχ––ΔσΤΎΝΌ¥≤ ‘―ι.Β¬ΙζHoeschst―–ΨΩΥυΒΡsaviprazole(HOE 731,7)Γ≤10Γ≥Μ·―ß–‘÷ Έ»Ε®Θ§Ψ≤¬ωΉΔ…δ“÷ΥαΜν–‘”κ(1c)œύΥΤΘ§Ε‘―Σ“ΚΈΗΟΎΥΊΒΡ≈®Ε»”Αœλ≤Μ¥σ.
1.3 »Γ¥ζΏδΏράύ
»’±ΨTanabe,SeiyakuΙΪΥΨ’ΐΕ‘¥ΥάύΜ·ΚœΈοΫχ––SAR―–ΨΩΘ§“―…Η―Γ≥ωT-330(8)ΚΆT-776(9)Γ≤11Γ≥Θ§≤Δ―–ΨΩΝΥ(9)Ε‘H+/K+-ATPΟΗΒΡΉς”ΟΜζ÷ΤΘΚ‘ΎΈΗ±ΎœΗΑϊΒΡΥα–‘ΜΖΨ≥÷–Θ§(9)Ζ÷Ή”÷–ΒΡS(O)ΓΣCH2Φϋ‘ΎΥα¥ΏΜ·œ¬ΕœΩΣΘ§–Έ≥…Μν–‘Μ·ΚœΈο¥ΈΜ«ΥαΘ§”κH+/K+-ATPΟΗΫαΚœ≥…Ar-S-S-Enzymeάύ–ΆΒΡΟΗ-“÷÷ΤΦΝΗ¥ΚœΈο.’β÷÷ΕΰΝρΦϋ≤ΜΈ»Ε®,Η¥ΚœΈο»ί“ΉΆ§ΡΎ‘¥–‘ΒΡέœΜυΜ·ΚœΈοΖ¥”ΠΘ§H+/K+-ATPΟΗΡήΫœΩλΒΊΜ÷Η¥Μν–‘Θ§“ρ¥Υ(9)Ε‘H+/K+-ATPΟΗΒΡ“÷÷Τ”–“ΜΕ®ΒΡΩ…Ρφ–‘.
, ΑΌΡ¥“Ϋ“©
1.4 ΡαΙ≈ΕΓθΘΑΖάύ
»’±ΨDainippon“Ϋ“©ΙΪΥΨΕ‘±Ϋ≤ΔΏδΏράύ“‘ΆβΒΡΜ·ΚœΈοΫχ––…Η―ΓΘ§ΖΔœ÷“λύγΏρΆΣ≤ΔΏΝύΛ―ή…ζΈο(10)Ε‘H+/K+-ATPΟΗ”–ΫœΗΏΒΡ―Γ‘ώ–‘.ΒΪ’β÷÷Μ·ΚœΈο‘ΎΕ·ΈοΧεΡΎ ‘―ι÷–ΟΜ”–“÷ΥαΉς”ΟΘ§¥Υ―–ΨΩΥυ…ηΦΤΚœ≥…ΝΥΤδ«Α“©2-(ή–Μυ―«Μ«θΘΜυ)ΡαΙ≈ΕΓθΘΑΖ―ή…ζΈοΘ§…Η―Γ≥ωAD-8717(11a)Γ≤12Γ≥ΚΆAD-9161(11b)Ϋχ“Μ≤Ϋ―–ΨΩ.(11a)≤Μ”κΤδΥϋΒΡΟΗΚΆ ήΧεΉς”ΟΘ§≤Μ“÷÷ΤΗΈ‘ύΈΔΝΘΧεœΗΑϊ…ΪΥΊP450ΟΗœΒΒΡ“©Έο¥ζ–ΜΉς”ΟΘ§Ε‘H+/K+-ATPΟΗ”–ΚήΗΏΒΡ―Γ‘ώ–‘ΘΜ“Μ¥ΈΗχ“©“÷ΟΗΉς”ΟΩ…≥÷–χ48 h,Ρή‘ΎΫœ≥ΛΒΡ ±ΦδΡΎ…ΐΗΏΈΗΡΎpH÷ΒΘ§’βΕ‘œϊ≥ΐ”ΡΟ≈¬ίΗΥΨζ”–άϊ.(11a)ΒΡ“÷ΥαΚΆΩΙάΘ―ώΉς”Ο «(1c)ΒΡ2ΓΪ3±Ε.‘ΎΦ±–‘ΕΨ–‘ ‘―ι÷–Θ§–Γ σΩΎΖΰ(11a)1000 mg/kgΈ¥ΦϊΕΨ–‘”Αœλ.(11a)‘ΎΒΆpHΧθΦΰœ¬Θ§ΉΣΜ·ΈΣ”–Μν–‘ΒΡ“λύγΏρΆΣ≤ΔΏΝύΛ―ή…ζΈοΘ§≤ΜΩ…ΡφΒΊ“÷÷ΤH+/K+-ATPΟΗΒΡΜν–‘ΘΜΒΪ‘Ύ»θΥα–‘ΧθΦΰœ¬≤Μ“ΉΉΣΜ·Θ§‘Ύ÷––‘ΜρΗϋΗΏpHΧθΦΰœ¬Θ§≤ΜΖΔ…ζΉΣΜ·Θ§≤ΜΜαΕ‘ΈΗΥαΙΐΕ»“÷÷Τ.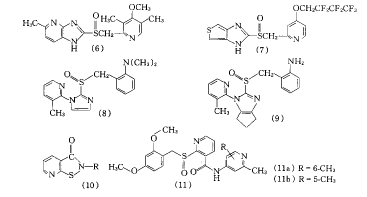
, ΑΌΡ¥“Ϋ“©
2 Ω…Ρφ–ΆPPI
Ω…Ρφ–ΆPPIΦ¥K+-όΉΩΙ–ΆPPI.±ΎœΗΑϊΖ÷ΟΎΙήΡΛ…œΒΡH+/K+-ATPΟΗ «“Μ÷÷ΩγΡΛΒΑΑΉΘ§Υϋ”–ΝΫΗωK+«ΉΚΆ≤ΩΈΜΘΚK+ΒΆ«ΉΚΆ≤ΩΈΜΘ§H+”κK+‘Ύ’βΗω≤ΩΈΜ…œΖΔ…ζΫΜΜΜ≤Δ±ΜΉΣ‘Υ(œΗΑϊΫ§÷–ΒΡH+Ϋχ»κΖ÷ΟΎΙήΘ§Ά§ ±Ζ÷ΟΎΙή÷–Β»ΝΩΒΡK+Ϋχ»κœΗΑϊΫ§)ΘΜK+ΗΏ«ΉΚΆ≤ΩΈΜΘ§ΟφœρΖ÷ΟΎΙή“Μ≤ύΘ§K+”κ¥ΥΈΜΒψΫαΚœΩ…ΦΛΜνH+/K+-ATPΟΗ.Ω…Ρφ–ΆPPIΕύ «»θΦν–‘‘”ΜΖάύΜ·ΚœΈοΘ§Ρή‘Ύ±ΎœΗΑϊΒΡΖ÷ΟΎΙή÷–ΖΔ…ζ÷ Ή”Μ·Θ§Τδ÷ Ή”Μ·Χε”κK+ΨΚ’υΗΏ«ΉΚΆ≤ΩΈΜΘ§“÷÷ΤH+/K+-ATPΟΗΒΡΜν–‘Θ§ ΙΈΗΥαΖ÷ΟΎ ήΒΫ“÷÷ΤΘΜ“ρK+‘ΎCl-±ΟΒΡΉς”Οœ¬≤ΜΕœΒΊ”…œΗΑϊΫ§ά©…ΔΒΫΖ÷ΟΎΙήΘ§Ζ÷ΟΎΙή÷–ΒΡK+≈®Ε»Μα÷πΫΞ…ΐΗΏΘ§K+”÷Ρή”κΤδΗΏ«ΉΚΆ≤ΩΈΜΫαΚœΘ§ ΙH+/K+-ATPΟΗΒΡΜν–‘Μ÷Η¥.
, ΑΌΡ¥“Ϋ“©
Ω…Ρφ–ΆPPIΕ‘H+/K+-ATPΟΗΒΡ“÷÷Τ «Ω…ΡφΒΡΘ§≥÷–χ ±ΦδΫœΕΧΘ§ΡήΫœΚΟΒΊΤπΒΫΒςΫΎΈΗΥαΖ÷ΟΎΒΡΉς”Ο.’ΐ‘Ύ―–ΨΩΒΡΩ…Ρφ–ΆPPI÷ς“Σ”–ΏδΏρ≤ΔΓ≤1,2-aΓ≥ΏΝύΛΓΔΑΖΜυύ≠ΏχΓΔύγΖ‘≤ΔΓ≤2,3-cΓ≥-1,5-ίΝύΛΓΔύγΖ‘≤ΔΓ≤3,2-eΓ≥±Ϋ≤ΔΏδΏρΓΔΏ…ΜυύγΏρΒ»―ή…ζΈο.
2.1 ΏδΏρ≤ΔΓ≤1,2-aΓ≥ΏΝύΛ―ή…ζΈο
1982ΡξΘ§ΟάΙζSchering-PloughΙΪΥΨ¥”“Μ–©ΏδΏρ≤ΔΓ≤1,2-aΓ≥ΏΝύΛΜ·ΚœΈο÷–…Η―Γ≥ωSCH-28080(12)Γ≤13Γ≥.ΥϋΦ»”–“÷ΥαΉς”ΟΘ§”÷”–ΈΗ’≥ΡΛœΗΑϊ±ΘΜΛΉς”ΟΘ§ΒΪΉν≥θΕ‘Τδ“÷ΥαΜζάμ≤Μ«ε≥ΰ.1986ΡξΘ§SewingΒ»»ΥΖΔœ÷Υϋ «“Μ÷÷–¬–ΆΒΡΓΔΩ…Ρφ–‘ΒΡPPIΘ§Φ¥K+-όΉΩΙ–ΆPPIΓ≤14Γ≥.(12)“ρΗΈΕΨ–‘Έ ΧβΈ¥Ρή”Ο”ΎΝΌ¥≤Θ§ΤδΕΨ–‘”κΤδ‘ΎΧεΡΎΒΡ¥ζ–Μ”–ΙΊΘ§¥ζ–ΜΒΡ≤ΩΈΜ‘ΎΜ·ΚœΈοΒΡ3-ΈΜΓΔ8-ΈΜΚΆΏΝύΛΜΖ…œΘ§7-ΈΜΕ‘¥ζ–Μ“≤”–”Αœλ.Ε‘¥ΥάύΜ·ΚœΈοΒΡΫαΙΙ-Μν–‘-ΕΨ–‘ΙΊœΒΫχ“Μ≤Ϋ―–ΨΩΘ§…Η―Γ≥ωΖϊΚœ“Σ«σΒΡΓΔΜν–‘”κ(12)œύΥΤΒΡΏδΏρ≤ΔΓ≤1,2-cΓ≥ΏΝύΚ―ή…ζΈοSCH-32651(13)Γ≤15Γ≥.»’±ΨYamanouchi“Ϋ“©ΙΪΥΨΒΡYM020(14)Γ≤16Γ≥Θ§“≤Φφ”–“÷÷ΤΈΗΥαΖ÷ΟΎΚΆΈΗ’≥ΡΛœΗΑϊ±ΘΜΛΉς”ΟΘ§“÷ΥαΜν–‘±»(1c)»θΘ§±»ΉιΑΖH2- ήΧεόΉΩΙΦΝΈςΟΉΧφΕΓ«ΩΘ§“÷ΥαΉς”Ο≥÷–χΒΡ ±ΦδΫœΕΧΘ§ΈόΟςœ‘Η±Ής”Ο.
, ΑΌΡ¥“Ϋ“©
¥ΥάύΜ·ΚœΈοΒΡpKa÷ΒΚΆΩ’ΦδΫαΙΙΒ»Ε‘Τδ“÷ΥαΜν–‘Ψυ”–”Αœλ.SCH-28080(12)ΒΡ8-ΈΜ»Γ¥ζΜυ”–ΝΫ÷÷Ω’Φδ»ΓœρΘΚ»Γ¥ζΜυΒΡ±ΫΜΖ’έœρΡΗΜΖ(heterocyclic nucleus)Θ§≥ΤΈΣΓΑ’έΒΰ ΫΓ±ΘΜ»Γ¥ζΜυΒΡ±ΫΜΖ‘ΕάκΡΗΜΖΘ§≥ΤΈΣΓΑ…λ’Ι ΫΓ±.άμ¬έΦΤΥψΦΑΈοάμ ΐΨί±μΟςΘ§ΓΑ…λ’Ι ΫΓ±±»ΓΑ’έΒΰ ΫΓ±Έ»Ε®.Ε‘8-±Ϋ““œ©ΜυΏδΏρ≤ΔΓ≤1,2-aΓ≥ΏΝύΛΆ§œΒΈοΒΡΙΙ–ßΙΊœΒ―–ΨΩΫαΙϊ“≤÷ΛΟςΘ§Μν–‘ΧεΈΣΓΑ…λ’Ι ΫΓ±ΫαΙΙ.Schering-PloughΙΪΥΨ»‘‘ΎΫχ––’βΖΫΟφΒΡ―–ΨΩΙΛΉςΓ≤17Γ≥.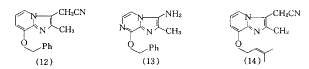
2.2 ΑΖΜυύ≠Ώχ―ή…ζΈο
Smith KlineΙΪΥΨ“‘4-ΖΦΑΖΜυύ≠Ώχ-3-θΞ(15)ΈΣœ»ΒΦΜ·ΚœΈοΘ§Ε‘Τδ3-ΈΜΚΆ8-ΈΜ»Γ¥ζΜυΫχ––ΫαΙΙ”≈Μ·Θ§…Η―Γ≥ωSK&F 96067(16a)Γ≤18Γ≥ΚΆSK&F 97574(16b)Γ≤19Γ≥.(16a)Ψ≤¬ωΉΔ…δΚΆΩΎΖΰΜν–‘ΨυΈΣΉιΑΖH2- ήΧεόΉΩΙΦΝΈςΟΉΧφΕΓΒΡ5ΓΪ6±ΕΘ§œϊ≥ΐΑκΥΞΤΎ≥Λ(16ΓΪ24 h)Θ§“÷ΥαΉς”Ο”κ―Σ“©≈®Ε»”–ΙΊΘ§÷ΈΝΤΦΝΝΩΈόΟςœ‘Η±Ής”ΟΘ§’ΐΫχ––ΔσΤΎΝΌ¥≤ ‘―ι.(16b)ΩΎΖΰΜν–‘‘ΦΈΣΈςΟΉΧφΕΓΒΡ10±ΕΘ§“÷ΥαΉς”Ο≥÷–χΒΡ ±Φδ±»(16a)≥ΛΘ§ΒΪ±»≤ΜΩ…Ρφ–ΆPPIΕΧΘ§’ΐΫχ––ΔώΤΎΝΌ¥≤ ‘―ι.»’±ΨOtsuka“Ϋ“©ΙΪΥΨΚœ≥…ΝΥ“ΜœΒΝ–4-ΖΦΑΖΜυύ≠Ώχ-3-θΘΑΖ―ή…ζΈοΓ≤20Γ≥Θ§Τδ÷–”–“Μ–©Μ·ΚœΈοΒΡΜν–‘ΫœΚΟΘ§»γ(16c)Β».(16c)Ε‘H+/K+-ATPΟΗΒΡ“÷÷Τ”κ―Σ“©≈®Ε»”–ΙΊΘ§‘ΎΥ°œύ÷–Έ»Ε®.Smith KlineΙΪΥΨ‘χ…ηΦΤΚœ≥…ΝΥ“ΜœΒΝ–ΏΝΩ©≤Δύ≠Ώχ―ή…ζΈοΓ≤21Γ≥Θ§―Γ»ΓSK&F 96356(17)―–ΨΩΝΥ¥ΥάύΜ·ΚœΈοΕ‘H+/K+-ATPΟΗΒΡΉς”ΟΜζ÷Τ.
, http://www.100md.com
2.3 ≥μ‘”≤ΔύΉύΛ―ή…ζΈο
Smith KlineΙΪΥΨΕ‘ύ≠ΏρΏχ―ή…ζΈο(18)ΚΆύγΖ‘≤ΔύΉύΛ―ή…ζΈο(19)Γ≤22Γ≥“≤ΉωΝΥ“Μ–©―–ΨΩΘ§ΖΔœ÷¥ΥάύΜ·ΚœΈοΒΡpKa÷ΒΚΆΩ’ΦδΫαΙΙΕ‘ΤδΜν–‘Ψυ”–”Αœλ.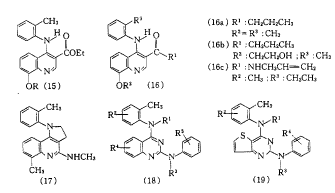
2.4 ΤδΥϋΒΡΩ…Ρφ–ΆPPI
ΤδΥϋ’ΐ‘Ύ―–ΨΩΒΡΩ…Ρφ–ΆPPI”–ύγΖ‘≤ΔΓ≤3,2-eΓ≥±Ϋ≤ΔΏδΏρ―ή…ζΈο(20)Γ≤23Γ≥ΓΔύγΖ‘≤ΔΓ≤2,3-cΓ≥-1,5-ίΝύΛ―ή…ζΈο(21)Γ≤24Γ≥ΓΔκ“ΜυύγΏρ―ή…ζΈο(22)Γ≤25Γ≥Β».(20)÷–Υδ”–“Μ–©Μ·ΚœΈοΜν–‘ΫœΚΟΘ§ΒΪΥϋΟ«‘ΎΫœ¥σΦΝΝΩ ±“ΐΤπ―œ÷ΊΒΡΗ±Ζ¥”ΠΘ§¥Υœ»ΒΦΜ·ΚœΈο–η“ΣΫχ“Μ≤ΫΫαΙΙ”≈Μ·.(21)ΒΡ4-ΈΜ»Γ¥ζΜυΈΣΑΖΜυ ±Μν–‘ΫœΚΟΘ§ΒΪ‘ΕΒΆ”Ύ(16a).Ε‘(22)Ϋχ––ΫαΙΙ–ό Έ“≤ΟΜ”–’“ΒΫάμœκΒΡΜν–‘Μ·ΚœΈο.
, ΑΌΡ¥“Ϋ“©
3 ΤδΥϋΒΡPPI
“Μ–©Χλ»Μ≤ζΈο»γΒΞΡΰΒ»Εύτ«ΜυΖ”―ή…ζΈοΓ≤26Γ≥Θ§Ε‘H+/K+-ATPΟΗ”–“÷÷ΤΉς”Ο.ΤδΉς”ΟΜζ÷ΤΈΣΕύτ«ΜυΖ”―ή…ζΈο”κATPΨΚ’υΈΜΒψΘ§Ήη÷ΙATP”κH+/K+-ATPΟΗΒΡΫαΚœΦΑΝΉΥαΜ·ΟΗΒΡ–Έ≥…Θ§¥”Εχ“÷÷ΤΟΗΒΡΜν–‘.
»’±ΨMeiji SeikaΙΪΥΨΩΣΖΔΒΡME-3407(23)Γ≤27Γ≥Θ§Ής”ΟΜζάμ≤ΜΆ§”ΎΤδΥϋΒΡPPIΘ§ΥϋΉηΕœH+/K+-ATPΟΗ¥”ΙήΉ¥Ρ“≈ίœρΖ÷ΟΎΙήΡΛΒΡΉΣ“ΤΘ§ Ι’β“Μ≤ΩΖ÷ΟΗ≤ΜΡήΖΔΜ”Ής”ΟΘ§ΒΪΕ‘Ζ÷ΟΎΙήΡΛ…œΒΡH+/K+-ATPΟΗΈό“÷÷ΤΉς”Ο.(23)Ω…”Ο”Ύ÷ΈΝΤΦ±–‘ΚΆ¬ΐ–‘ΈΗάΘ―ώΘ§Τδ÷–ΩΎΖΰΉν”––ßΘ§’ΐΫχ––ΔσΤΎΝΌ¥≤ ‘―ι.ΝμΆβ“Μ–©Μ·ΚœΈο»γWY-26769(24)Θ§AY-28200(25)ΚΆΜ·ΚœΈο(26)Β»Θ§‘χ±®ΒάΤδΕ‘H+/K+-ATPΟΗΒΡ“÷÷ΤΉς”ΟΘ§ΒΪΉς”ΟΜζ÷Τ…–≤Μ«ε≥ΰ.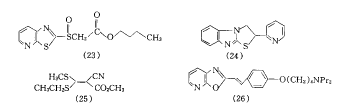
, ΑΌΡ¥“Ϋ“©
Ήή÷°Θ§PPI÷–“―”–4Ηω–¬“©±Μ≈ζΉΦ…œ –Θ§÷Ν…Ό”–9ΗωΜ·ΚœΈο’ΐ‘ΎΫχ––ΝΌ¥≤―–ΨΩ.PPI“÷÷ΤΈΗΥαΖ÷ΟΎΒΡΉνΚσ“ΜΗωΜΖΫΎΘ§ΡήΙΜ“÷÷ΤΗς÷÷“ρΥΊ“ΐΤπΒΡΈΗΥαΖ÷ΟΎΘ§”Ο”Ύ÷ΈΝΤΈΗάΘ―ώΓΔ °Εΰ÷Η≥ΠάΘ―ώ ±Θ§Οςœ‘”≈”ΎΉιΑΖH2- ήΧεόΉΩΙΦΝ.“―≈ζΉΦ…œ –ΒΡPPIΨυΈΣ≤ΜΩ…Ρφ–ΆΒΡ±Ϋ≤ΔΏδΏράύΜ·ΚœΈοΘ§ΥφΉ≈ΥϋΟ«‘ΎΝΌ¥≤…œΒΡΙψΖΚ”Π”ΟΘ§ΥϋΟ«ΒΡ»±Βψ÷πΫΞ±μœ÷≥ωά¥Θ§»γ“÷ΟΗΉς”ΟΙΐ«ΩΓΔ≥÷–χ ±ΦδΧΪ≥ΛΓΔ≥ΛΤΎ”Ο“©“Ή“ΐΤπΈΗΥα»±ΖΠ÷ΔΒ».ΈΗΥα»±ΖΠ ±Θ§―Σ“Κ÷–ΈΗΟΎΥΊ≈®Ε»Μα…ΐΗΏΘΜΈΗΥα―œ÷Ί»±ΖΠ ±Θ§”–“ΐΤπ≥Π »ΗθœΗΑϊ‘ω…ζΓΔΒΦ÷¬ΈΗΑ©ΒΡΈΘœ’.≤ΜΩ…Ρφ–ΆPPI÷–Θ§”––©Μ·ΚœΈο“ρΤδΕάΧΊΒΡ“©¥ζΕ·ΝΠ―ßΧΊΒψΘ§“÷÷ΤΈΗΥαΖ÷ΟΎΒΡ≥ΧΕ»≤Μ «ΧΪ…νΘ§Ε‘―Σ“ΚΈΗΟΎΥΊΒΡ≈®Ε»”Αœλ≤Μ¥σΘ§ «ΡΩ«Α―–ΨΩΒΡ“ΜΗωΖΫœρ.ΒΪ≤ΜΩ…Ρφ–ΆPPIΕ‘H+/K+-ATPΟΗΒΡ“÷÷Τ «≤ΜΩ…Ρφ–‘ΒΡΘ§’β÷÷“÷÷Τ≤ΜΡήΥφΉ≈ΈΗΥαΖ÷ΟΎΒΡ±δΜ·ΦΑ ±Ήω≥ωΖ¥”ΠΘ§ΟΗΒΡΜν–‘Μ÷Η¥±»Ϋœ≥ΌΜΚΘ§”ΑœλΟΗΒΡ’ΐ≥ΘΙΠΡήΒΡΖΔΜ”.K+-όΉΩΙ–ΆPPIΆ®Ιΐ”κK+ΨΚ’υΗΏ«ΉΚΆ≤ΩΈΜΘ§“÷÷ΤH+/K+-ATPΟΗΒΡΜν–‘Θ§’β÷÷“÷÷Τ «Ω…Ρφ–‘ΒΡΘ§Β±ΈΗΥαΖ÷ΟΎ±Μ“÷÷ΤΒΫ“ΜΕ®ΒΡ≥ΧΕ» ±Θ§ΟΗΒΡΜν–‘ΜαΥφΉ≈±ΎœΗΑϊΖ÷ΟΎΙή÷–K+≈®Ε»ΒΡ…ΐΗΏΕχΜ÷Η¥Θ§≤ΜΜαΙΐΕ»“÷÷ΤΈΗΥαΖ÷ΟΎΘ§”κ≤ΜΩ…Ρφ–ΆPPI±»ΫœΘ§ΫœΈΣάμœκΘ§ «ΡΩ«Α―–ΨΩΒΡ“ΜΗω÷Ί“ΣΖΫœρ.ΝμΆβΘ§Ε‘H+/K+-ATPΟΗΒΡ―–ΨΩΫχ’Ι“≤±»ΫœΩλΘ§“―±®ΒάΝΥΥϋΒΡ»ΐΈ§ΫαΙΙΓ≤28Γ≥Θ§’βΈΣ”Π”ΟΦΤΥψΜζ“©ΈοΗ®÷ζ…ηΦΤ―Α’“–¬ΒΡPPIΒλΕ®ΝΥΜυ¥ΓΘ§ΫΪΜα¥ΌΫχPPIΒΡΩΣΖΔΫχ≥Χ.
, ΑΌΡ¥“Ϋ“©
≤ΈΩΦΈΡœΉ
1Θ§Ganser AL,Forte JG.K+-stimulated ATPase in purified microsomes of bullfrog oxyntic cells.Biochim Biophys Acta,1973,304:169ΓΪ180
2Θ§Fellenius E,Berglindh T,Sachs G,et al.Substituted benzimidazoles inhibitor gastric acid secretion by blocking H+/K+-ATPase.Nature,1981,290(5802):159ΓΪ161
3Θ§Lindberg P,Nordberg P,Alminger T,et al.The mechanism of action of the gastric acid secretion inhibitor omeprazole.J Med Chem,1986,29(8):1327ΓΪ1329
, ΑΌΡ¥“Ϋ“©
4Θ§Kim PY,Jung WT,Lee SM.IY-81149.Drugs Fut,1999,24(6):618ΓΪ621
5Θ§Yamada S,Goto T,Yamaguchi T,et al.Synthetic study of 2-Γ≤(6,7,8,9-tetrahydro-5H-cycloheptaΓ≤bΓ≥pyridin-9-yl)-sulfinylΓ≥-1H-benzimidazole analogs and their biological properties as novel proton pump inhibitors.Chem Pharm Bull,1995,43(3):421ΓΪ431
6Θ§Terashima K,Shimamura H,Kawase A,et al.Studies on antiulcer agents.1.Synthesis and pharmacological properties of ethyl 2-Γ≤(1H-benzimidazol-2-yl)sulfinylmethylΓ≥-4-dimethylamino-5-pyrimidinecarboxylate,a new H+/K+-ATPase inhibitor possessing mucosal protective activity.Chem Pharm Bull,1995,43(1):166ΓΪ168
, ΑΌΡ¥“Ϋ“©
7Θ§Ishihara K,Ichikawa T,Kmuro Y,et al.Effect on gastric mucus of the proton pump inhibitor leminoprazole and its cytoprotective action against ethanol-induced gastric injury in rats.Arzneim-Forsch,1994,44(7):823ΓΪ830
8Θ§Uchida M,Chihiro M,Morita S,et al.Synthesis and antiulcer activity of 4-substituted 8-Γ≤(2-benzimidazolyl)sulfinylmethylΓ≥-1,2,3,4-tetrahydroquinolines and related compounds.Chem Pharm Bull,1990,38(6):1575ΓΪ1586
9Θ§Prous J,Mealy N,Castaner J.TU-199.Drugs Fut,1994,19(11):1018ΓΪ1019
, ΑΌΡ¥“Ϋ“©
10Θ§Weidmann K,Herling AW,Lang HJ,et al.2-Γ≤(2-pyridylmethyl)sulfinylΓ≥-1H-thienoΓ≤3,4-dΓ≥imidazoles,a novel class of gastric H+/K+-ATPase inhibitors.J Med Chem,1992,35(3):438ΓΪ450
11Θ§Yamada M,Yura T,Morimoto M,et al.2-Γ≤(2-aminobenzyl)sulfinylΓ≥-1-(2-pyridyl)-1,4,5,6-tetrahydrocyclopentΓ≤dΓ≥imidazoles as a novel class of gastric H+/K+-ATPase inhibitors.J Med Chem,1996,39(2):596ΓΪ604
12Θ§Nakamura K,Terauchi H,Komiya M,et al.AD-8717.Drugs Fut,1997,22(12):1309ΓΪ1313
, ΑΌΡ¥“Ϋ“©
13Θ§Kaminski JJ,Bristol JA,Puchalski C,et al.Antiulcer agents.1.Gastric antisecretory and cytoprotective properties of substituted imidazoΓ≤1,2-aΓ≥pyridines.J Med Chem,1985,28(7):876ΓΪ892
14Θ§Beil W,Hackbarth I,Sewing KF.Mechanism of gastric antisecretory effects of SCH-28080.Br J Pharmacol,1986,88(1):19ΓΪ23
15Θ§Kaminski JJ,Perkins DG,Frantz JD,et al.Antiulcer agents.3.Structure-activity-toxicity relationship of substituted imidazoΓ≤1,2-aΓ≥pyridines and a related imidazoΓ≤1,2-aΓ≥pyrazine.J Med Chem,1987,30(11):2047ΓΪ2051
, ΑΌΡ¥“Ϋ“©
16Θ§Yuki H,Kamato T,Nishida A,et al.Antisecretory and antiulcer effects of YM020,a new H+/K+-ATPase inhibitor,in rats and dogs.Jpn J Pharmacol,1995,67(1):59ΓΪ67
17Θ§Kaminski JJ,Doweyko AM.Antiulcer agents.6.Analysis of the in vitro biochemical and in vivo gastric antisecertory activity of substituted imidazoΓ≤1,2-aΓ≥pyridines and related analogues using comparative molecular field analysis and hypothetical active site lattice methodologies.J Med Chem,1997,40(4):427ΓΪ436
, ΑΌΡ¥“Ϋ“©
18Θ§Ife RJ,Brown TH,Keeling DJ,et al.Reversible inhibitors of the gastric H+/K+-ATPase.3.3-Substituted-4-(phenylamino)quinolines.J Med Chem,1992,35(18):3413ΓΪ3422
19Θ§Leach CA,Brown TH,Ife RJ,et al.Reversible inhibitors of the gastric H+/K+-ATPase.4.Identification of an inhibitor with an intermediate duration of action.J Med Chem,1995,38(14):2748ΓΪ2762
20Θ§Uchida M,Otsrbo K,Matsubara J,et al.Synthesis of 4-(phenylamino)quinoline-3-carboxamides as a novel class of gastric H+/K+-ATPase inhibitors.Chem Pharm Bull,1995,43(4):693ΓΪ698
, ΑΌΡ¥“Ϋ“©
21Θ§Leach CA,Brown TH,Ife RJ,et al.Reversible inhibitors of the gastric H+/K+-ATPase.2.1-ArylpyrroloΓ≤3,2-cΓ≥quinolines:effect of the 4-substitutent.J Med Chem,1992,35(10):1845ΓΪ1852
22Θ§Ife RJ,Brown TH,Blurton P,et al.Reversible inhibitors of the gastric H+/K+-ATPase.5.Substituted 2,4-diaminoquinazolines and thienopyrimidines.J Med Chem,1995,38(14):2763ΓΪ2773
23Θ§Homma K,Watanabe T,Iijima T,et al.Efficient synthesis and gastric H+/K+-ATPase inhibitory activity of 2-aryl-4,5-dihydro-1H-thienoΓ≤3,2-eΓ≥benzimidazoles.Chem Pharm Bull,1997,45(12):1945ΓΪ1954
, ΑΌΡ¥“Ϋ“©
24Θ§Bjork P,Hornfeldt AB,Gronowitz S,et al.4-Substituted 1-(2-methylphenyl)thienoΓ≤2,3-cΓ≥-1,5-naphthyridines as possible reversible inhibitors of gastric H+/K+-ATPase.Eur J Med Chem,1996,31(5):411ΓΪ416
25Θ§LaMattina JL,McCarthy PA,Reiter LA,et al.Antiulcer agents.4-substituted 2-guanidinothiazoles:reversible,competitive,and selective inhibitors of gastric H+/K+-ATPase.J Med Chem,1990,33(2):543ΓΪ552
, http://www.100md.com
26Θ§Murakami S,Muramatsu M,Otomo S.Inhibitory effect of tannica acid on gastric H+/K+-ATPase.J Nat Prod,1992,55(4):513ΓΪ516
27Θ§Prous J,Mealy N,Castaner J.ME-3407.Drugs Fut,1994,19(1):31ΓΪ32
28Θ§Xian YJ,Hebert H.Three-dimensional structure of the porcine gastric H+/K+-ATPase from negatively stained crystals.J Stuct Biol,1997,118:169ΓΪ177
’Ηε»’ΤΎ:2000-02-26, ΑΌΡ¥“Ϋ“©
ΒΞΈΜΘΚ…ρ―τ“©ΩΤ¥σ―ß“©ΈοΚœ≥… “Θ§…ρ―τ 110015
ΙΊΦϋ¥ ΘΚH+/K+-ATPΟΗ“÷÷ΤΦΝΘΜ“÷Υα“©ΈοΘΜΩΙάΘ―ώΘΜΩ…Ρφ–ΆΘΜ≤ΜΩ…Ρφ–ΆΘΜΉς”ΟΜζάμ
÷–Ιζ“©ΈοΜ·―ß‘”÷Ψ000222 ’Σ“Σ Ϋι…ήΝΥ“÷Υα“©ΈοH+/K+-ATPΟΗ“÷÷ΤΦΝΒΡ―–ΨΩ«ιΩωΚΆΖΔ’ΙΖΫœρΘ§≤ΔΕ‘“―Ψ≠±®ΒάΒΡH+/K+-ATPΟΗ“÷÷ΤΦΝΒΡΉς”ΟΜζάμΫχ––ΝΥ±»Ϋœ.
Advance on the Research and Development of H+/K+-ATPase Inhibitors
Zhai Jianguo,Yan Dong,Cheng Maosheng
, http://www.100md.com
(Laboratory of Drug Synthesis,Shenyang Pharmaceutical University,Shenyang 110015)
Abstract This is about advance on the research and development of H+/K+-ATPase inhibitors.The mechanism of action of H+/K+-ATPase inhibitors was also discussed.
Key words H+/K+TPase inhibitors;antisecretory;antiulcer agents;reversible;irreversible;mechanism
, ΑΌΡ¥“Ϋ“©
H+/K+-ATPΟΗ“÷÷ΤΦΝΘ§Φ¥÷ Ή”±Ο“÷÷ΤΦΝ(proton pump inhibitors,PPI), «“Μάύ–¬–ΆΒΡ“÷÷ΤΈΗΥαΖ÷ΟΎΒΡ“©Έο.H+/K+-ATPΟΗ(÷ Ή”±Ο)Θ§ΈΜ”Ύ±ΎœΗΑϊΒΡΙήΉ¥Ρ“≈ίΚΆΖ÷ΟΎΙήΡΛ…œΘ§≤Έ”κΈΗΥαΖ÷ΟΎΒΡΉνΚσ“ΜΗωΜΖΫΎΘ§“÷÷Τ¥ΥΟΗΒΡΜν–‘Θ§Ω…“÷÷ΤΗς÷÷“ρΥΊ“ΐΤπΒΡΈΗΥαΖ÷ΟΎ.PPI÷ς“Σ”Ο”Ύ÷ΈΝΤΈΗάΘ―ώΓΔ °Εΰ÷Η≥ΠάΘ―ώΓΔΖ¥Νς–‘ ≥Ιή―ΉΚΆΉτ-ΌΙ œΉέΚœ÷ΔΒ»”κΈΗΥαΖ÷ΟΎ ßΒς”–ΙΊΒΡΦ≤≤Γ.’βάύ“©ΈοΫœΉιΑΖH2- ήΧεόΉΩΙΦΝΦΑΤδΥϋ“÷÷ΤΈΗΥαΖ÷ΟΎΒΡ“©ΈοΘ§ΨΏ”–Οςœ‘ΒΡ”≈‘Ϋ–‘Θ§»γ―Γ‘ώ–‘ΗΏΓΔΝΤ–ßΚΟΓΔΗ±Ής”Ο…ΌΓΔ”κΩΙ…ζΥΊ≈δΈιΒΡΗ¥ΖΫ÷ΤΦΝΩ…œϊ≥ΐ”ΡΟ≈¬ίΗΥΨζ(ΈΗάΘ―ώ÷¬≤ΓΨζ)Β».PPI“―”–4Ηω–¬“©≈ζΉΦ…œ –Θ§’ΐ‘ΎΜρΦ¥ΫΪΫχ––ΝΌ¥≤ ‘―ιΒΡ“≤”–≤Μ…Ό.ΗυΨίΤδ”κH+/K+-ATPΟΗΉς”ΟΒΡ≤ΜΆ§ΖΫ ΫΘ§÷ς“ΣΖ÷ΈΣΩ…Ρφ–ΆΚΆ≤ΜΩ…Ρφ–ΆΝΫάύ.ΡΩ«ΑPPIΒΡ―–ΨΩΕύΈΣ―Α’“Ω…Ρφ–ΆΜν–‘“©Έο.
1 ≤ΜΩ…Ρφ–ΆPPI
, http://www.100md.com
≤ΜΩ…Ρφ–ΆPPI «“Μ÷÷«Α“©Θ§Υϋ‘ΎΈΗ±ΎœΗΑϊΒΡΥα–‘ΜΖΨ≥÷–ΉΣΜ·ΈΣ¥ΈΜ«ΥαΜν–‘ΧεΘ§”κH+/K+-ATPΟΗ“‘ΕΰΝρΦϋΫαΚœΘ§–Έ≥…ΟΗ-“÷÷ΤΦΝΗ¥ΚœΈοΘ§≤ΜΩ…ΡφΒΊ“÷÷ΤH+/K+-ATPΟΗΒΡΜν–‘Θ§ΫΒΒΆΈΗΥαΖ÷ΟΎ.’ΐ‘Ύ―–ΨΩΒΡ≤ΜΩ…Ρφ–ΆPPI÷ς“Σ”–±Ϋ≤ΔΏδΏράύΓΔ≥μ‘”≤ΔΏδΏράύΓΔ»Γ¥ζΏδΏράύΚΆΡαΙ≈ΕΓθΘΑΖάύΒ»―ή…ζΈο.
1.1 ±Ϋ≤ΔΏδΏράύ
¥ΥάύPPIΒΡΜν–‘œ»ΒΦΜ·ΚœΈοtimeprazole(1a)Θ§ «»πΒδAstra Hassle―–ΨΩΥυ”Ύ1978Ρξ‘Ύ“÷Υα“©ΈοΒΡ…Η―Γ÷–ΖΔœ÷ΒΡΘ§“ρΒ± ±ForteΒ»»Υ“―ΖΔœ÷ΝΥH+/K+-ATPΟΗΓ≤1Γ≥Θ§‘χΆΤ≤β(1a)ΒΡ“÷ΥαΜζάμΈΣ“÷÷ΤH+/K+-ATPΟΗΒΡΜν–‘.÷°Κσ¥Υ―–ΨΩΥυΚœ≥…ΝΥpicoprazole(1b)ΚΆomeprazole(1c)Γ≤2,3Γ≥Θ§≤Δ≤ϊΟςΤδΕ‘H+/K+-ATPΟΗΒΡΉς”ΟΜζ÷Τ.(1c)”Ύ1988Ρξ≈ζΉΦ…œ –“‘ΚσΘ§“Μ÷±ΈΜ”Ύ άΫγ≥©œζ“©ΈοΒΡ«ΑΝ–Θ§ΤδΒΞ“ΜΒΡΉσ–ΐΙβ―ß“λΙΙΧεperprazole“―Ϋχ»κΔσΤΎΝΌ¥≤ ‘―ιΚσΤΎΘ§≤Δ‘ΎΟάΙζΜώΒΟ÷ΤΦΝΉ®άϊ.»’±ΨTakedaΙΪΥΨΒΡlansoprazole(1d)Θ§Β¬ΙζBykGuldenΙΪΥΨΒΡpantoprazole(1e)ΡΤ―ΈΚΆ»’±ΨEisaiΙΪΥΨΒΡrabeprazole(E3810,1f)ΡΤ―ΈΘ§Ζ÷±π”Ύ1991Θ§1994ΚΆ1997Ρξ≈ζΉΦ…œ –.ΚΪΙζΔρ-Yang“Ϋ“©ΙΪΥΨΒΡIY-81149(1g)Γ≤4Γ≥Θ§“÷÷Τ”ΡΟ≈¬ίΗΥΨζΒΡΉς”Ο«Ω”Ύ(1c)Θ§ΑκΥΞΤΎΕΧΘ§Ε‘―Σ“ΚΈΗΟΎΥΊΒΡ≈®Ε»”ΑœλΫœ–ΓΘ§ΩΙΈΗάΘ―ώΉς”Ο «(1c)ΒΡ3±ΕΘ§«“≤Μ≤ζ…ζΩΙ“©–‘Θ§ΈόΟςœ‘Η±Ής”ΟΘ§Α≤»Ϊ–‘ΫœΚΟΘ§’ΐΫχ––ΔρΤΎΝΌ¥≤ ‘―ι.ΨΏ”–(1a)Μυ±ΨΫαΙΙΒΡPPIΜΙ”–ΚήΕύ’ΐ‘ΎΩΣΖΔΙΐ≥Χ÷–Θ§»γdisuprazole(1h),SK&F-95601(1i)ΚΆRo-18-5364(1j)Β».
, ΑΌΡ¥“Ϋ“©
Ε‘œ»ΒΦΜ·ΚœΈο(1a)ΒΡΫαΙΙ–ό Έ”–“ΜΕ®ΒΡΨ÷œό–‘Θ§–μΕύΙΪΥΨΕ‘±Ϋ≤ΔΏδΏρΜΖΚΆΏΝύΛΜΖΒΡΩ…Χφ¥ζ–‘Ϋχ––ΝΥ―–ΨΩΘ§ΖΔœ÷ΝΥ–μΕύ“÷ΥαΚΆΩΙάΘ―ώΉς”ΟΕΦΚήΚΟΒΡPPI.»γ»’±ΨToaEiyoΙΪΥΨΒΡTY-11345(2)Γ≤5Γ≥Θ§“÷ΥαΉς”Ο «(1c)ΒΡ2ΓΪ3±ΕΘ§ΩΙάΘ―ώΉς”Ο «(1c)ΒΡ3ΓΪ15±ΕΘ§’ΐΫχ––ΔρΤΎΝΌ¥≤ ‘―ι.ύΉύΛΦΉΥαθΞ―ή…ζΈο(3)Γ≤6Γ≥ΩΎΖΰΚΆΨ≤¬ωΉΔ…δΨυ”–ΈΗ’≥ΡΛœΗΑϊ±ΘΜΛΉς”ΟΘ§ΤδΩΙάΘ―ώΉς”Ο «ΉιΑΖH2- ήΧεόΉΩΙΦΝΈςΟΉΧφΕΓΒΡ3±Ε.±ΫΑΖ―ή…ζΈοNC-1300(4a)ΚΆNC-1300-O-3(leminoprazole,4b)Γ≤7Γ≥Θ§Φφ”–“÷÷ΤΈΗΥαΖ÷ΟΎΚΆΈΗ’≥ΡΛœΗΑϊ±ΘΜΛΉς”Ο.(4b)”…»’±ΨNippon ChemipharΙΪΥΨ―–÷ΤΘ§ΥϋΒΡΩΙάΘ―ώΜν–‘”κ(1c)œύΥΤΘ§Ε‘H+/K+-ATPΟΗΒΡ“÷÷Τ≥÷–χ ±ΦδΫœ≥ΛΘ§”–ΩΙ”ΡΟ≈¬ίΗΥΨζΉς”ΟΘ§Ε‘÷– ύΚΆΉ‘÷ς…ώΨ≠œΒΆ≥ΓΔ–Ρ―ΣΙήœΒΆ≥Β»ΈόΟςœ‘”ΑœλΘ§’ΐΫχ––ΔσΤΎΝΌ¥≤ ‘―ι.“―±®ΒάΒΡΜΙ”–S-3337(4c)Β».1,2,3,4-ΥΡ«βύ≠Ώχ―ή…ζΈοOPC-22381(5a)ΚΆOPC-22575(5b)Γ≤8Γ≥Β»Θ§“÷ΥαΉς”ΟΫœ»θ.
, http://www.100md.com
±Ϋ≤ΔΏδΏράύPPIΒΡ¥ζ±μΜ·ΚœΈο «(1c)Θ§ΥϋΫχ»κΈΗΒΡ±ΎœΗΑϊΚσΘ§‘ΎΖ÷ΟΎΙή÷–H+ΒΡΉς”Οœ¬Θ§ΖΔ…ζΖ÷Ή”ΡΎ«ΉΚΥΖ¥”ΠΘ§–Έ≥…¬ί÷–ΦδΧεΘ§ΉΣΜ·≥…¥ΈΜ«ΥαΓΔ¥ΈΜ«θΘΑΖΜν–‘Μ·ΚœΈοΘ§”κH+/K+-ATPΟΗΠΝ―«Μυ÷–ΒΡCys813ΚΆCys892≤–Μυ“‘ΕΰΝρΦϋΫαΚœΘ§–Έ≥…ΟΗ-“÷÷ΤΦΝΗ¥ΚœΈοΘ§“÷÷ΤH+/K+-ATPΟΗΒΡΜν–‘.ΤδΥϋΒΡ±Ϋ≤ΔΏδΏράύPPIΕ‘H+/K+-ATPΟΗΒΡΉς”ΟΜζ÷ΤάύΥΤ”Ύ(1c)Θ§“≤ «”κΡ≥Cys≤–ΜυΫαΚœ.
±Ϋ≤ΔΏδΏράύPPIΕ‘H+/K+-ATPΟΗΒΡ“÷÷ΤΉς”ΟΫœ«ΩΘ§Εχ«“≥÷ΨΟΘ§Ε‘ΈΗΥα“÷÷ΤΒΡ≥ΧΕ»Ϋœ…νΘ§“ΜΑψΕΧΤΎ”Ο“©ΨΆΡή÷Έ”ζΈΗάΘ―ώΜρ °Εΰ÷Η≥ΠάΘ―ώΘΜΒΪ≥ΛΤΎ”Ο“©Θ§“Ή“ΐΤπ”κΈΗΥα»±ΖΠ”–ΙΊΒΡΗ±Ζ¥”ΠΘ§»γœϊΜ·≤ΜΝΦΓΔΗΏΈΗΟΎΥΊ―Σ÷ΔΓΔ≥Π »ΗθœΗΑϊ‘ω…ζΒ».≤ΩΖ÷≤Γ»Υ”Ο“©Κσ≥ωœ÷Εώ–ΡΓΔ≈ΜΆ¬ΓΔ’ΆΤχΓΔΗΙ–ΚΒ»≤ΜΝΦΖ¥”ΠΘ§“ΜΑψ≤Μ–ηΦθΝΩΜρΆΘ÷Ι÷ΈΝΤ.≥ΐ(1e)ΆβΘ§±Ϋ≤ΔΏδΏράύPPIΜΙΡή“÷÷ΤΗΈΈΔΝΘΧεœΗΑϊ…ΪΥΊP450ΟΗœΒΒΡ“©Έο¥ζ–ΜΉς”ΟΘ§ΫΒΒΆœψΕΙΥΊάύΩΙΡΐ“©ΓΔΑ≤Ε®ΓΔ±ΫΆΉ“ρΒ»“©ΈοΒΡ«ε≥ΐ¬ .
, http://www.100md.com
1.2 ≥μ‘”≤ΔΏδΏράύ
»’±ΨTokyo TanabeΙΪΥΨΒΡTU-199(6)Γ≤9Γ≥“÷ΥαΚΆΩΙάΘ―ώΉς”ΟΕΦ≥§Ιΐ(1c)Θ§«“Μ·―ßΈ»Ε®–‘ΚΟΘ§’ΐΫχ––ΔσΤΎΝΌ¥≤ ‘―ι.Β¬ΙζHoeschst―–ΨΩΥυΒΡsaviprazole(HOE 731,7)Γ≤10Γ≥Μ·―ß–‘÷ Έ»Ε®Θ§Ψ≤¬ωΉΔ…δ“÷ΥαΜν–‘”κ(1c)œύΥΤΘ§Ε‘―Σ“ΚΈΗΟΎΥΊΒΡ≈®Ε»”Αœλ≤Μ¥σ.
1.3 »Γ¥ζΏδΏράύ
»’±ΨTanabe,SeiyakuΙΪΥΨ’ΐΕ‘¥ΥάύΜ·ΚœΈοΫχ––SAR―–ΨΩΘ§“―…Η―Γ≥ωT-330(8)ΚΆT-776(9)Γ≤11Γ≥Θ§≤Δ―–ΨΩΝΥ(9)Ε‘H+/K+-ATPΟΗΒΡΉς”ΟΜζ÷ΤΘΚ‘ΎΈΗ±ΎœΗΑϊΒΡΥα–‘ΜΖΨ≥÷–Θ§(9)Ζ÷Ή”÷–ΒΡS(O)ΓΣCH2Φϋ‘ΎΥα¥ΏΜ·œ¬ΕœΩΣΘ§–Έ≥…Μν–‘Μ·ΚœΈο¥ΈΜ«ΥαΘ§”κH+/K+-ATPΟΗΫαΚœ≥…Ar-S-S-Enzymeάύ–ΆΒΡΟΗ-“÷÷ΤΦΝΗ¥ΚœΈο.’β÷÷ΕΰΝρΦϋ≤ΜΈ»Ε®,Η¥ΚœΈο»ί“ΉΆ§ΡΎ‘¥–‘ΒΡέœΜυΜ·ΚœΈοΖ¥”ΠΘ§H+/K+-ATPΟΗΡήΫœΩλΒΊΜ÷Η¥Μν–‘Θ§“ρ¥Υ(9)Ε‘H+/K+-ATPΟΗΒΡ“÷÷Τ”–“ΜΕ®ΒΡΩ…Ρφ–‘.
, ΑΌΡ¥“Ϋ“©
1.4 ΡαΙ≈ΕΓθΘΑΖάύ
»’±ΨDainippon“Ϋ“©ΙΪΥΨΕ‘±Ϋ≤ΔΏδΏράύ“‘ΆβΒΡΜ·ΚœΈοΫχ––…Η―ΓΘ§ΖΔœ÷“λύγΏρΆΣ≤ΔΏΝύΛ―ή…ζΈο(10)Ε‘H+/K+-ATPΟΗ”–ΫœΗΏΒΡ―Γ‘ώ–‘.ΒΪ’β÷÷Μ·ΚœΈο‘ΎΕ·ΈοΧεΡΎ ‘―ι÷–ΟΜ”–“÷ΥαΉς”ΟΘ§¥Υ―–ΨΩΥυ…ηΦΤΚœ≥…ΝΥΤδ«Α“©2-(ή–Μυ―«Μ«θΘΜυ)ΡαΙ≈ΕΓθΘΑΖ―ή…ζΈοΘ§…Η―Γ≥ωAD-8717(11a)Γ≤12Γ≥ΚΆAD-9161(11b)Ϋχ“Μ≤Ϋ―–ΨΩ.(11a)≤Μ”κΤδΥϋΒΡΟΗΚΆ ήΧεΉς”ΟΘ§≤Μ“÷÷ΤΗΈ‘ύΈΔΝΘΧεœΗΑϊ…ΪΥΊP450ΟΗœΒΒΡ“©Έο¥ζ–ΜΉς”ΟΘ§Ε‘H+/K+-ATPΟΗ”–ΚήΗΏΒΡ―Γ‘ώ–‘ΘΜ“Μ¥ΈΗχ“©“÷ΟΗΉς”ΟΩ…≥÷–χ48 h,Ρή‘ΎΫœ≥ΛΒΡ ±ΦδΡΎ…ΐΗΏΈΗΡΎpH÷ΒΘ§’βΕ‘œϊ≥ΐ”ΡΟ≈¬ίΗΥΨζ”–άϊ.(11a)ΒΡ“÷ΥαΚΆΩΙάΘ―ώΉς”Ο «(1c)ΒΡ2ΓΪ3±Ε.‘ΎΦ±–‘ΕΨ–‘ ‘―ι÷–Θ§–Γ σΩΎΖΰ(11a)1000 mg/kgΈ¥ΦϊΕΨ–‘”Αœλ.(11a)‘ΎΒΆpHΧθΦΰœ¬Θ§ΉΣΜ·ΈΣ”–Μν–‘ΒΡ“λύγΏρΆΣ≤ΔΏΝύΛ―ή…ζΈοΘ§≤ΜΩ…ΡφΒΊ“÷÷ΤH+/K+-ATPΟΗΒΡΜν–‘ΘΜΒΪ‘Ύ»θΥα–‘ΧθΦΰœ¬≤Μ“ΉΉΣΜ·Θ§‘Ύ÷––‘ΜρΗϋΗΏpHΧθΦΰœ¬Θ§≤ΜΖΔ…ζΉΣΜ·Θ§≤ΜΜαΕ‘ΈΗΥαΙΐΕ»“÷÷Τ.
, ΑΌΡ¥“Ϋ“©
2 Ω…Ρφ–ΆPPI
Ω…Ρφ–ΆPPIΦ¥K+-όΉΩΙ–ΆPPI.±ΎœΗΑϊΖ÷ΟΎΙήΡΛ…œΒΡH+/K+-ATPΟΗ «“Μ÷÷ΩγΡΛΒΑΑΉΘ§Υϋ”–ΝΫΗωK+«ΉΚΆ≤ΩΈΜΘΚK+ΒΆ«ΉΚΆ≤ΩΈΜΘ§H+”κK+‘Ύ’βΗω≤ΩΈΜ…œΖΔ…ζΫΜΜΜ≤Δ±ΜΉΣ‘Υ(œΗΑϊΫ§÷–ΒΡH+Ϋχ»κΖ÷ΟΎΙήΘ§Ά§ ±Ζ÷ΟΎΙή÷–Β»ΝΩΒΡK+Ϋχ»κœΗΑϊΫ§)ΘΜK+ΗΏ«ΉΚΆ≤ΩΈΜΘ§ΟφœρΖ÷ΟΎΙή“Μ≤ύΘ§K+”κ¥ΥΈΜΒψΫαΚœΩ…ΦΛΜνH+/K+-ATPΟΗ.Ω…Ρφ–ΆPPIΕύ «»θΦν–‘‘”ΜΖάύΜ·ΚœΈοΘ§Ρή‘Ύ±ΎœΗΑϊΒΡΖ÷ΟΎΙή÷–ΖΔ…ζ÷ Ή”Μ·Θ§Τδ÷ Ή”Μ·Χε”κK+ΨΚ’υΗΏ«ΉΚΆ≤ΩΈΜΘ§“÷÷ΤH+/K+-ATPΟΗΒΡΜν–‘Θ§ ΙΈΗΥαΖ÷ΟΎ ήΒΫ“÷÷ΤΘΜ“ρK+‘ΎCl-±ΟΒΡΉς”Οœ¬≤ΜΕœΒΊ”…œΗΑϊΫ§ά©…ΔΒΫΖ÷ΟΎΙήΘ§Ζ÷ΟΎΙή÷–ΒΡK+≈®Ε»Μα÷πΫΞ…ΐΗΏΘ§K+”÷Ρή”κΤδΗΏ«ΉΚΆ≤ΩΈΜΫαΚœΘ§ ΙH+/K+-ATPΟΗΒΡΜν–‘Μ÷Η¥.
, ΑΌΡ¥“Ϋ“©
Ω…Ρφ–ΆPPIΕ‘H+/K+-ATPΟΗΒΡ“÷÷Τ «Ω…ΡφΒΡΘ§≥÷–χ ±ΦδΫœΕΧΘ§ΡήΫœΚΟΒΊΤπΒΫΒςΫΎΈΗΥαΖ÷ΟΎΒΡΉς”Ο.’ΐ‘Ύ―–ΨΩΒΡΩ…Ρφ–ΆPPI÷ς“Σ”–ΏδΏρ≤ΔΓ≤1,2-aΓ≥ΏΝύΛΓΔΑΖΜυύ≠ΏχΓΔύγΖ‘≤ΔΓ≤2,3-cΓ≥-1,5-ίΝύΛΓΔύγΖ‘≤ΔΓ≤3,2-eΓ≥±Ϋ≤ΔΏδΏρΓΔΏ…ΜυύγΏρΒ»―ή…ζΈο.
2.1 ΏδΏρ≤ΔΓ≤1,2-aΓ≥ΏΝύΛ―ή…ζΈο
1982ΡξΘ§ΟάΙζSchering-PloughΙΪΥΨ¥”“Μ–©ΏδΏρ≤ΔΓ≤1,2-aΓ≥ΏΝύΛΜ·ΚœΈο÷–…Η―Γ≥ωSCH-28080(12)Γ≤13Γ≥.ΥϋΦ»”–“÷ΥαΉς”ΟΘ§”÷”–ΈΗ’≥ΡΛœΗΑϊ±ΘΜΛΉς”ΟΘ§ΒΪΉν≥θΕ‘Τδ“÷ΥαΜζάμ≤Μ«ε≥ΰ.1986ΡξΘ§SewingΒ»»ΥΖΔœ÷Υϋ «“Μ÷÷–¬–ΆΒΡΓΔΩ…Ρφ–‘ΒΡPPIΘ§Φ¥K+-όΉΩΙ–ΆPPIΓ≤14Γ≥.(12)“ρΗΈΕΨ–‘Έ ΧβΈ¥Ρή”Ο”ΎΝΌ¥≤Θ§ΤδΕΨ–‘”κΤδ‘ΎΧεΡΎΒΡ¥ζ–Μ”–ΙΊΘ§¥ζ–ΜΒΡ≤ΩΈΜ‘ΎΜ·ΚœΈοΒΡ3-ΈΜΓΔ8-ΈΜΚΆΏΝύΛΜΖ…œΘ§7-ΈΜΕ‘¥ζ–Μ“≤”–”Αœλ.Ε‘¥ΥάύΜ·ΚœΈοΒΡΫαΙΙ-Μν–‘-ΕΨ–‘ΙΊœΒΫχ“Μ≤Ϋ―–ΨΩΘ§…Η―Γ≥ωΖϊΚœ“Σ«σΒΡΓΔΜν–‘”κ(12)œύΥΤΒΡΏδΏρ≤ΔΓ≤1,2-cΓ≥ΏΝύΚ―ή…ζΈοSCH-32651(13)Γ≤15Γ≥.»’±ΨYamanouchi“Ϋ“©ΙΪΥΨΒΡYM020(14)Γ≤16Γ≥Θ§“≤Φφ”–“÷÷ΤΈΗΥαΖ÷ΟΎΚΆΈΗ’≥ΡΛœΗΑϊ±ΘΜΛΉς”ΟΘ§“÷ΥαΜν–‘±»(1c)»θΘ§±»ΉιΑΖH2- ήΧεόΉΩΙΦΝΈςΟΉΧφΕΓ«ΩΘ§“÷ΥαΉς”Ο≥÷–χΒΡ ±ΦδΫœΕΧΘ§ΈόΟςœ‘Η±Ής”Ο.
, ΑΌΡ¥“Ϋ“©
¥ΥάύΜ·ΚœΈοΒΡpKa÷ΒΚΆΩ’ΦδΫαΙΙΒ»Ε‘Τδ“÷ΥαΜν–‘Ψυ”–”Αœλ.SCH-28080(12)ΒΡ8-ΈΜ»Γ¥ζΜυ”–ΝΫ÷÷Ω’Φδ»ΓœρΘΚ»Γ¥ζΜυΒΡ±ΫΜΖ’έœρΡΗΜΖ(heterocyclic nucleus)Θ§≥ΤΈΣΓΑ’έΒΰ ΫΓ±ΘΜ»Γ¥ζΜυΒΡ±ΫΜΖ‘ΕάκΡΗΜΖΘ§≥ΤΈΣΓΑ…λ’Ι ΫΓ±.άμ¬έΦΤΥψΦΑΈοάμ ΐΨί±μΟςΘ§ΓΑ…λ’Ι ΫΓ±±»ΓΑ’έΒΰ ΫΓ±Έ»Ε®.Ε‘8-±Ϋ““œ©ΜυΏδΏρ≤ΔΓ≤1,2-aΓ≥ΏΝύΛΆ§œΒΈοΒΡΙΙ–ßΙΊœΒ―–ΨΩΫαΙϊ“≤÷ΛΟςΘ§Μν–‘ΧεΈΣΓΑ…λ’Ι ΫΓ±ΫαΙΙ.Schering-PloughΙΪΥΨ»‘‘ΎΫχ––’βΖΫΟφΒΡ―–ΨΩΙΛΉςΓ≤17Γ≥.
2.2 ΑΖΜυύ≠Ώχ―ή…ζΈο
Smith KlineΙΪΥΨ“‘4-ΖΦΑΖΜυύ≠Ώχ-3-θΞ(15)ΈΣœ»ΒΦΜ·ΚœΈοΘ§Ε‘Τδ3-ΈΜΚΆ8-ΈΜ»Γ¥ζΜυΫχ––ΫαΙΙ”≈Μ·Θ§…Η―Γ≥ωSK&F 96067(16a)Γ≤18Γ≥ΚΆSK&F 97574(16b)Γ≤19Γ≥.(16a)Ψ≤¬ωΉΔ…δΚΆΩΎΖΰΜν–‘ΨυΈΣΉιΑΖH2- ήΧεόΉΩΙΦΝΈςΟΉΧφΕΓΒΡ5ΓΪ6±ΕΘ§œϊ≥ΐΑκΥΞΤΎ≥Λ(16ΓΪ24 h)Θ§“÷ΥαΉς”Ο”κ―Σ“©≈®Ε»”–ΙΊΘ§÷ΈΝΤΦΝΝΩΈόΟςœ‘Η±Ής”ΟΘ§’ΐΫχ––ΔσΤΎΝΌ¥≤ ‘―ι.(16b)ΩΎΖΰΜν–‘‘ΦΈΣΈςΟΉΧφΕΓΒΡ10±ΕΘ§“÷ΥαΉς”Ο≥÷–χΒΡ ±Φδ±»(16a)≥ΛΘ§ΒΪ±»≤ΜΩ…Ρφ–ΆPPIΕΧΘ§’ΐΫχ––ΔώΤΎΝΌ¥≤ ‘―ι.»’±ΨOtsuka“Ϋ“©ΙΪΥΨΚœ≥…ΝΥ“ΜœΒΝ–4-ΖΦΑΖΜυύ≠Ώχ-3-θΘΑΖ―ή…ζΈοΓ≤20Γ≥Θ§Τδ÷–”–“Μ–©Μ·ΚœΈοΒΡΜν–‘ΫœΚΟΘ§»γ(16c)Β».(16c)Ε‘H+/K+-ATPΟΗΒΡ“÷÷Τ”κ―Σ“©≈®Ε»”–ΙΊΘ§‘ΎΥ°œύ÷–Έ»Ε®.Smith KlineΙΪΥΨ‘χ…ηΦΤΚœ≥…ΝΥ“ΜœΒΝ–ΏΝΩ©≤Δύ≠Ώχ―ή…ζΈοΓ≤21Γ≥Θ§―Γ»ΓSK&F 96356(17)―–ΨΩΝΥ¥ΥάύΜ·ΚœΈοΕ‘H+/K+-ATPΟΗΒΡΉς”ΟΜζ÷Τ.
, http://www.100md.com
2.3 ≥μ‘”≤ΔύΉύΛ―ή…ζΈο
Smith KlineΙΪΥΨΕ‘ύ≠ΏρΏχ―ή…ζΈο(18)ΚΆύγΖ‘≤ΔύΉύΛ―ή…ζΈο(19)Γ≤22Γ≥“≤ΉωΝΥ“Μ–©―–ΨΩΘ§ΖΔœ÷¥ΥάύΜ·ΚœΈοΒΡpKa÷ΒΚΆΩ’ΦδΫαΙΙΕ‘ΤδΜν–‘Ψυ”–”Αœλ.
2.4 ΤδΥϋΒΡΩ…Ρφ–ΆPPI
ΤδΥϋ’ΐ‘Ύ―–ΨΩΒΡΩ…Ρφ–ΆPPI”–ύγΖ‘≤ΔΓ≤3,2-eΓ≥±Ϋ≤ΔΏδΏρ―ή…ζΈο(20)Γ≤23Γ≥ΓΔύγΖ‘≤ΔΓ≤2,3-cΓ≥-1,5-ίΝύΛ―ή…ζΈο(21)Γ≤24Γ≥ΓΔκ“ΜυύγΏρ―ή…ζΈο(22)Γ≤25Γ≥Β».(20)÷–Υδ”–“Μ–©Μ·ΚœΈοΜν–‘ΫœΚΟΘ§ΒΪΥϋΟ«‘ΎΫœ¥σΦΝΝΩ ±“ΐΤπ―œ÷ΊΒΡΗ±Ζ¥”ΠΘ§¥Υœ»ΒΦΜ·ΚœΈο–η“ΣΫχ“Μ≤ΫΫαΙΙ”≈Μ·.(21)ΒΡ4-ΈΜ»Γ¥ζΜυΈΣΑΖΜυ ±Μν–‘ΫœΚΟΘ§ΒΪ‘ΕΒΆ”Ύ(16a).Ε‘(22)Ϋχ––ΫαΙΙ–ό Έ“≤ΟΜ”–’“ΒΫάμœκΒΡΜν–‘Μ·ΚœΈο.
, ΑΌΡ¥“Ϋ“©
3 ΤδΥϋΒΡPPI
“Μ–©Χλ»Μ≤ζΈο»γΒΞΡΰΒ»Εύτ«ΜυΖ”―ή…ζΈοΓ≤26Γ≥Θ§Ε‘H+/K+-ATPΟΗ”–“÷÷ΤΉς”Ο.ΤδΉς”ΟΜζ÷ΤΈΣΕύτ«ΜυΖ”―ή…ζΈο”κATPΨΚ’υΈΜΒψΘ§Ήη÷ΙATP”κH+/K+-ATPΟΗΒΡΫαΚœΦΑΝΉΥαΜ·ΟΗΒΡ–Έ≥…Θ§¥”Εχ“÷÷ΤΟΗΒΡΜν–‘.
»’±ΨMeiji SeikaΙΪΥΨΩΣΖΔΒΡME-3407(23)Γ≤27Γ≥Θ§Ής”ΟΜζάμ≤ΜΆ§”ΎΤδΥϋΒΡPPIΘ§ΥϋΉηΕœH+/K+-ATPΟΗ¥”ΙήΉ¥Ρ“≈ίœρΖ÷ΟΎΙήΡΛΒΡΉΣ“ΤΘ§ Ι’β“Μ≤ΩΖ÷ΟΗ≤ΜΡήΖΔΜ”Ής”ΟΘ§ΒΪΕ‘Ζ÷ΟΎΙήΡΛ…œΒΡH+/K+-ATPΟΗΈό“÷÷ΤΉς”Ο.(23)Ω…”Ο”Ύ÷ΈΝΤΦ±–‘ΚΆ¬ΐ–‘ΈΗάΘ―ώΘ§Τδ÷–ΩΎΖΰΉν”––ßΘ§’ΐΫχ––ΔσΤΎΝΌ¥≤ ‘―ι.ΝμΆβ“Μ–©Μ·ΚœΈο»γWY-26769(24)Θ§AY-28200(25)ΚΆΜ·ΚœΈο(26)Β»Θ§‘χ±®ΒάΤδΕ‘H+/K+-ATPΟΗΒΡ“÷÷ΤΉς”ΟΘ§ΒΪΉς”ΟΜζ÷Τ…–≤Μ«ε≥ΰ.
, ΑΌΡ¥“Ϋ“©
Ήή÷°Θ§PPI÷–“―”–4Ηω–¬“©±Μ≈ζΉΦ…œ –Θ§÷Ν…Ό”–9ΗωΜ·ΚœΈο’ΐ‘ΎΫχ––ΝΌ¥≤―–ΨΩ.PPI“÷÷ΤΈΗΥαΖ÷ΟΎΒΡΉνΚσ“ΜΗωΜΖΫΎΘ§ΡήΙΜ“÷÷ΤΗς÷÷“ρΥΊ“ΐΤπΒΡΈΗΥαΖ÷ΟΎΘ§”Ο”Ύ÷ΈΝΤΈΗάΘ―ώΓΔ °Εΰ÷Η≥ΠάΘ―ώ ±Θ§Οςœ‘”≈”ΎΉιΑΖH2- ήΧεόΉΩΙΦΝ.“―≈ζΉΦ…œ –ΒΡPPIΨυΈΣ≤ΜΩ…Ρφ–ΆΒΡ±Ϋ≤ΔΏδΏράύΜ·ΚœΈοΘ§ΥφΉ≈ΥϋΟ«‘ΎΝΌ¥≤…œΒΡΙψΖΚ”Π”ΟΘ§ΥϋΟ«ΒΡ»±Βψ÷πΫΞ±μœ÷≥ωά¥Θ§»γ“÷ΟΗΉς”ΟΙΐ«ΩΓΔ≥÷–χ ±ΦδΧΪ≥ΛΓΔ≥ΛΤΎ”Ο“©“Ή“ΐΤπΈΗΥα»±ΖΠ÷ΔΒ».ΈΗΥα»±ΖΠ ±Θ§―Σ“Κ÷–ΈΗΟΎΥΊ≈®Ε»Μα…ΐΗΏΘΜΈΗΥα―œ÷Ί»±ΖΠ ±Θ§”–“ΐΤπ≥Π »ΗθœΗΑϊ‘ω…ζΓΔΒΦ÷¬ΈΗΑ©ΒΡΈΘœ’.≤ΜΩ…Ρφ–ΆPPI÷–Θ§”––©Μ·ΚœΈο“ρΤδΕάΧΊΒΡ“©¥ζΕ·ΝΠ―ßΧΊΒψΘ§“÷÷ΤΈΗΥαΖ÷ΟΎΒΡ≥ΧΕ»≤Μ «ΧΪ…νΘ§Ε‘―Σ“ΚΈΗΟΎΥΊΒΡ≈®Ε»”Αœλ≤Μ¥σΘ§ «ΡΩ«Α―–ΨΩΒΡ“ΜΗωΖΫœρ.ΒΪ≤ΜΩ…Ρφ–ΆPPIΕ‘H+/K+-ATPΟΗΒΡ“÷÷Τ «≤ΜΩ…Ρφ–‘ΒΡΘ§’β÷÷“÷÷Τ≤ΜΡήΥφΉ≈ΈΗΥαΖ÷ΟΎΒΡ±δΜ·ΦΑ ±Ήω≥ωΖ¥”ΠΘ§ΟΗΒΡΜν–‘Μ÷Η¥±»Ϋœ≥ΌΜΚΘ§”ΑœλΟΗΒΡ’ΐ≥ΘΙΠΡήΒΡΖΔΜ”.K+-όΉΩΙ–ΆPPIΆ®Ιΐ”κK+ΨΚ’υΗΏ«ΉΚΆ≤ΩΈΜΘ§“÷÷ΤH+/K+-ATPΟΗΒΡΜν–‘Θ§’β÷÷“÷÷Τ «Ω…Ρφ–‘ΒΡΘ§Β±ΈΗΥαΖ÷ΟΎ±Μ“÷÷ΤΒΫ“ΜΕ®ΒΡ≥ΧΕ» ±Θ§ΟΗΒΡΜν–‘ΜαΥφΉ≈±ΎœΗΑϊΖ÷ΟΎΙή÷–K+≈®Ε»ΒΡ…ΐΗΏΕχΜ÷Η¥Θ§≤ΜΜαΙΐΕ»“÷÷ΤΈΗΥαΖ÷ΟΎΘ§”κ≤ΜΩ…Ρφ–ΆPPI±»ΫœΘ§ΫœΈΣάμœκΘ§ «ΡΩ«Α―–ΨΩΒΡ“ΜΗω÷Ί“ΣΖΫœρ.ΝμΆβΘ§Ε‘H+/K+-ATPΟΗΒΡ―–ΨΩΫχ’Ι“≤±»ΫœΩλΘ§“―±®ΒάΝΥΥϋΒΡ»ΐΈ§ΫαΙΙΓ≤28Γ≥Θ§’βΈΣ”Π”ΟΦΤΥψΜζ“©ΈοΗ®÷ζ…ηΦΤ―Α’“–¬ΒΡPPIΒλΕ®ΝΥΜυ¥ΓΘ§ΫΪΜα¥ΌΫχPPIΒΡΩΣΖΔΫχ≥Χ.
, ΑΌΡ¥“Ϋ“©
≤ΈΩΦΈΡœΉ
1Θ§Ganser AL,Forte JG.K+-stimulated ATPase in purified microsomes of bullfrog oxyntic cells.Biochim Biophys Acta,1973,304:169ΓΪ180
2Θ§Fellenius E,Berglindh T,Sachs G,et al.Substituted benzimidazoles inhibitor gastric acid secretion by blocking H+/K+-ATPase.Nature,1981,290(5802):159ΓΪ161
3Θ§Lindberg P,Nordberg P,Alminger T,et al.The mechanism of action of the gastric acid secretion inhibitor omeprazole.J Med Chem,1986,29(8):1327ΓΪ1329
, ΑΌΡ¥“Ϋ“©
4Θ§Kim PY,Jung WT,Lee SM.IY-81149.Drugs Fut,1999,24(6):618ΓΪ621
5Θ§Yamada S,Goto T,Yamaguchi T,et al.Synthetic study of 2-Γ≤(6,7,8,9-tetrahydro-5H-cycloheptaΓ≤bΓ≥pyridin-9-yl)-sulfinylΓ≥-1H-benzimidazole analogs and their biological properties as novel proton pump inhibitors.Chem Pharm Bull,1995,43(3):421ΓΪ431
6Θ§Terashima K,Shimamura H,Kawase A,et al.Studies on antiulcer agents.1.Synthesis and pharmacological properties of ethyl 2-Γ≤(1H-benzimidazol-2-yl)sulfinylmethylΓ≥-4-dimethylamino-5-pyrimidinecarboxylate,a new H+/K+-ATPase inhibitor possessing mucosal protective activity.Chem Pharm Bull,1995,43(1):166ΓΪ168
, ΑΌΡ¥“Ϋ“©
7Θ§Ishihara K,Ichikawa T,Kmuro Y,et al.Effect on gastric mucus of the proton pump inhibitor leminoprazole and its cytoprotective action against ethanol-induced gastric injury in rats.Arzneim-Forsch,1994,44(7):823ΓΪ830
8Θ§Uchida M,Chihiro M,Morita S,et al.Synthesis and antiulcer activity of 4-substituted 8-Γ≤(2-benzimidazolyl)sulfinylmethylΓ≥-1,2,3,4-tetrahydroquinolines and related compounds.Chem Pharm Bull,1990,38(6):1575ΓΪ1586
9Θ§Prous J,Mealy N,Castaner J.TU-199.Drugs Fut,1994,19(11):1018ΓΪ1019
, ΑΌΡ¥“Ϋ“©
10Θ§Weidmann K,Herling AW,Lang HJ,et al.2-Γ≤(2-pyridylmethyl)sulfinylΓ≥-1H-thienoΓ≤3,4-dΓ≥imidazoles,a novel class of gastric H+/K+-ATPase inhibitors.J Med Chem,1992,35(3):438ΓΪ450
11Θ§Yamada M,Yura T,Morimoto M,et al.2-Γ≤(2-aminobenzyl)sulfinylΓ≥-1-(2-pyridyl)-1,4,5,6-tetrahydrocyclopentΓ≤dΓ≥imidazoles as a novel class of gastric H+/K+-ATPase inhibitors.J Med Chem,1996,39(2):596ΓΪ604
12Θ§Nakamura K,Terauchi H,Komiya M,et al.AD-8717.Drugs Fut,1997,22(12):1309ΓΪ1313
, ΑΌΡ¥“Ϋ“©
13Θ§Kaminski JJ,Bristol JA,Puchalski C,et al.Antiulcer agents.1.Gastric antisecretory and cytoprotective properties of substituted imidazoΓ≤1,2-aΓ≥pyridines.J Med Chem,1985,28(7):876ΓΪ892
14Θ§Beil W,Hackbarth I,Sewing KF.Mechanism of gastric antisecretory effects of SCH-28080.Br J Pharmacol,1986,88(1):19ΓΪ23
15Θ§Kaminski JJ,Perkins DG,Frantz JD,et al.Antiulcer agents.3.Structure-activity-toxicity relationship of substituted imidazoΓ≤1,2-aΓ≥pyridines and a related imidazoΓ≤1,2-aΓ≥pyrazine.J Med Chem,1987,30(11):2047ΓΪ2051
, ΑΌΡ¥“Ϋ“©
16Θ§Yuki H,Kamato T,Nishida A,et al.Antisecretory and antiulcer effects of YM020,a new H+/K+-ATPase inhibitor,in rats and dogs.Jpn J Pharmacol,1995,67(1):59ΓΪ67
17Θ§Kaminski JJ,Doweyko AM.Antiulcer agents.6.Analysis of the in vitro biochemical and in vivo gastric antisecertory activity of substituted imidazoΓ≤1,2-aΓ≥pyridines and related analogues using comparative molecular field analysis and hypothetical active site lattice methodologies.J Med Chem,1997,40(4):427ΓΪ436
, ΑΌΡ¥“Ϋ“©
18Θ§Ife RJ,Brown TH,Keeling DJ,et al.Reversible inhibitors of the gastric H+/K+-ATPase.3.3-Substituted-4-(phenylamino)quinolines.J Med Chem,1992,35(18):3413ΓΪ3422
19Θ§Leach CA,Brown TH,Ife RJ,et al.Reversible inhibitors of the gastric H+/K+-ATPase.4.Identification of an inhibitor with an intermediate duration of action.J Med Chem,1995,38(14):2748ΓΪ2762
20Θ§Uchida M,Otsrbo K,Matsubara J,et al.Synthesis of 4-(phenylamino)quinoline-3-carboxamides as a novel class of gastric H+/K+-ATPase inhibitors.Chem Pharm Bull,1995,43(4):693ΓΪ698
, ΑΌΡ¥“Ϋ“©
21Θ§Leach CA,Brown TH,Ife RJ,et al.Reversible inhibitors of the gastric H+/K+-ATPase.2.1-ArylpyrroloΓ≤3,2-cΓ≥quinolines:effect of the 4-substitutent.J Med Chem,1992,35(10):1845ΓΪ1852
22Θ§Ife RJ,Brown TH,Blurton P,et al.Reversible inhibitors of the gastric H+/K+-ATPase.5.Substituted 2,4-diaminoquinazolines and thienopyrimidines.J Med Chem,1995,38(14):2763ΓΪ2773
23Θ§Homma K,Watanabe T,Iijima T,et al.Efficient synthesis and gastric H+/K+-ATPase inhibitory activity of 2-aryl-4,5-dihydro-1H-thienoΓ≤3,2-eΓ≥benzimidazoles.Chem Pharm Bull,1997,45(12):1945ΓΪ1954
, ΑΌΡ¥“Ϋ“©
24Θ§Bjork P,Hornfeldt AB,Gronowitz S,et al.4-Substituted 1-(2-methylphenyl)thienoΓ≤2,3-cΓ≥-1,5-naphthyridines as possible reversible inhibitors of gastric H+/K+-ATPase.Eur J Med Chem,1996,31(5):411ΓΪ416
25Θ§LaMattina JL,McCarthy PA,Reiter LA,et al.Antiulcer agents.4-substituted 2-guanidinothiazoles:reversible,competitive,and selective inhibitors of gastric H+/K+-ATPase.J Med Chem,1990,33(2):543ΓΪ552
, http://www.100md.com
26Θ§Murakami S,Muramatsu M,Otomo S.Inhibitory effect of tannica acid on gastric H+/K+-ATPase.J Nat Prod,1992,55(4):513ΓΪ516
27Θ§Prous J,Mealy N,Castaner J.ME-3407.Drugs Fut,1994,19(1):31ΓΪ32
28Θ§Xian YJ,Hebert H.Three-dimensional structure of the porcine gastric H+/K+-ATPase from negatively stained crystals.J Stuct Biol,1997,118:169ΓΪ177
’Ηε»’ΤΎ:2000-02-26, ΑΌΡ¥“Ϋ“©