缺血性脑卒中的病理生理及药物治疗现状
作者:冯亦璞
单位:(中国医学科学院、中国协和医科大学药物研究所,北京 100050)
关键词:
药学学报990117.htm 目前脑血管病已成为我国3大病死原因之一[1]。对脑卒中病理生理的深入研究加深了对其预防和治疗的认识,并推动和加速对新型抗脑缺血药物的研制开发。
脑卒中大体可分成两大类:(1) 缺血性脑卒中,约占(70~80)%;(2) 出血性脑卒中,约占(20~30)%。这两类病变在治疗上完全不同。本文着重介绍缺血性脑卒中的病理生理和药物治疗。
1 脑缺血病理生理改变的因素[2]
虽然近年来,由于钙超载,毒性氧自由基和兴奋毒等学说的出现,为解释脑缺血的病理生理奠定了基础,一些治疗脑缺血的药物如钙通道阻滞药、自由基清除剂和兴奋性氨基酸(EAA)拮抗剂也应运而生,但迄今尚无较理想的治疗药物。因此,对脑缺血病理生理的深入研究仍是艰巨任务。Siesjo[3]认为全脑缺血时钙通过谷氨酸受体门控性和电压依赖性通道进入胞内。钙超载后触发了一系列影响再灌期神经元存活的反应,引起神经元存活后数小时乃至数日后继发性死亡。始初的损伤可能引起细胞膜钙处理能力的持续变化,导致线粒体持续、缓慢的钙超载。另一种可能是细胞内的信号转导通路持续紊乱而引起转录和翻译水平上的改变,及某些细胞(如产生应激蛋白的细胞,产生营养因子或对存活必需酶的细胞)的丢失。而在这一变化过程中,并未见微血管或线粒体功能障碍。与此相反,局部脑缺血虽然也会引起钙超载而触发一系列反应,但对脑细胞死亡的作用是次要的,而起主要作用的是在继发性损伤中产生的调质所引起的反应。这些调质可能是花生四烯酸(AA)代谢物,NO,自由基或其它活性代谢物。在缺血期或缺血1~3 h后的再灌期,这些调质可诱导内皮细胞和多形核白细胞(PMNLs)上粘连分子的表达或氧化关键蛋白质,还可触发炎性细胞因子如白介素(IL)-1,-6,-8,γ-干扰素,或肿瘤坏死因子(TNF)的合成和释放,引起缺血阴影区微血管阻塞。此外可产生由于持续缺血所引起的线粒体功能衰竭。因此局部脑缺血时微循环和线粒体功能衰竭是引起缺血继发性损伤的主要原因。近年来,在对继发性脑损伤中细胞因子和炎症反应所起的作用也已受到重视,以下就脑缺血后病理生理改变的多种因素,特别是对近年来一些新的看法作重点介绍。
, 百拇医药
细胞凋亡[4] 是指细胞按自身程序主动死亡的过程,而细胞坏死则是被动死亡过程。细胞凋亡在形态学和生化特徵上表现为细胞皱缩,细胞核染色质浓缩,DNA片断化,而细胞的膜结构和细胞器仍完整。研究发现,脑缺血后迟发性神经元损伤过程出现细胞凋亡,有人认为迟发性神经元损伤主要是由细胞凋亡所致,但对此仍有不同看法。在缺血缺氧时有几种因素调节细胞的生、死命运。一般认为,在抑制细胞凋亡过程有信号转导,如神经营养因子和应激蛋白等,它们分别激活抑制凋亡的基因如bcl2/bclXL等的表达,使细胞凋亡受到抑制,从而改善脑损伤;而在促进细胞凋亡过程中出现bax/bad和p53等基因的表达,激活某些蛋白水解酶,使修复DNA的酶如PARP(poly(ADP-ribose)polymerase)和DNA-PKcs(DNA-dependent protein kinase)等降解,最后导至细胞凋亡。由此可见,探讨凋亡与相关基因或相关因素(线粒体功能受损如ATP合成受阻、膜电位失衡、氧化损伤以及蛋白水解酶的激活等)之间的关系是很重要的,这为寻找治疗脑损伤药物提供线索。
, 百拇医药
自由基 脑缺血再灌期,当细胞内Ca2+浓度升高时,胞内依赖于Ca2+的蛋白水解酶被激活,黄嘌呤脱氢酶转变为黄嘌呤氧化酶(XO)。与此同时,ATP降解代谢物如次黄嘌呤(HPY)和黄嘌呤(Xan)增加。在XO作用下,产生大量毒性氧自由基。此外,花生四烯酸(AA)经环氧酶代谢途径产生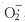 ,在铁蛋白存在下,转变为.OH,攻击膜结构和DNA,使细胞组织结构和功能均遭到破坏。此外,NO途径所产生的氧自由基也起重要作用[5]。在生理情况下,NO与的反应速度比超氧物歧化酶(SOD)清除的速度快3倍,但由于体内NO浓度(10~100 nmol.L-1)比体内SOD的浓度大约低100倍,此时产生的NO很难与SOD竞争,而主要作为信使传导分子行使其功能。在病理情况下如脑缺血/再灌时,细胞内NO的浓度显著升高,以至于达到能与SOD竞争超氧化物的水平,因为NO含有一个未配对电子,有存在时,迅速与其反应生成硝基过氧化物(ONOO—),后者质子化后进一步生成过氧亚硝酸(ONOOH),并在酸性环境中进一步分解为.OH和NO2。过去认为,.OH是NO引起脑损伤的最重要的氧自由基,现在则认为硝基过氧化物的直接氧化作用的毒性远大于.OH的毒性,因为硝基过氧化物不仅是一种很强的氧化剂,更重要的是它对反应有很高的选择性,如过氧化亚硝基能够直接氧化脂质、DNA以及蛋白质的巯基、锌/硫中心及铁/硫中心等,上述反应的速率大约是过氧化亚硝酸分解产生.OH速率的1 000倍。由此可见,硝基过氧化物可能是NO途径中产生的一种非常重要的氧自由基,以致严重损伤神经元。有关从抗氧自由基的途径去研究抗脑缺血药物也受到重视。据报道,维生素E和21位氨基类固醇化合物U74006F(商品名tirilazad)具有强的抗实验性脑缺血作用。对U74006F进行了多中心、随机、双盲和安慰剂对照临床试验,于脑卒中后4.3 h给药(6 mg.kg.d-1×3),未见神经功能改善[6]。
,在铁蛋白存在下,转变为.OH,攻击膜结构和DNA,使细胞组织结构和功能均遭到破坏。此外,NO途径所产生的氧自由基也起重要作用[5]。在生理情况下,NO与的反应速度比超氧物歧化酶(SOD)清除的速度快3倍,但由于体内NO浓度(10~100 nmol.L-1)比体内SOD的浓度大约低100倍,此时产生的NO很难与SOD竞争,而主要作为信使传导分子行使其功能。在病理情况下如脑缺血/再灌时,细胞内NO的浓度显著升高,以至于达到能与SOD竞争超氧化物的水平,因为NO含有一个未配对电子,有存在时,迅速与其反应生成硝基过氧化物(ONOO—),后者质子化后进一步生成过氧亚硝酸(ONOOH),并在酸性环境中进一步分解为.OH和NO2。过去认为,.OH是NO引起脑损伤的最重要的氧自由基,现在则认为硝基过氧化物的直接氧化作用的毒性远大于.OH的毒性,因为硝基过氧化物不仅是一种很强的氧化剂,更重要的是它对反应有很高的选择性,如过氧化亚硝基能够直接氧化脂质、DNA以及蛋白质的巯基、锌/硫中心及铁/硫中心等,上述反应的速率大约是过氧化亚硝酸分解产生.OH速率的1 000倍。由此可见,硝基过氧化物可能是NO途径中产生的一种非常重要的氧自由基,以致严重损伤神经元。有关从抗氧自由基的途径去研究抗脑缺血药物也受到重视。据报道,维生素E和21位氨基类固醇化合物U74006F(商品名tirilazad)具有强的抗实验性脑缺血作用。对U74006F进行了多中心、随机、双盲和安慰剂对照临床试验,于脑卒中后4.3 h给药(6 mg.kg.d-1×3),未见神经功能改善[6]。
, http://www.100md.com
一氧化氮[7~10] 它是一个能扩散的有高度化学活性的气体分子,半衰期短,可在细胞间自由地传递信使。而与经典的神经介质不同,它能在神经元之间或神经元与胶质细胞之间进行顺向和逆向的信息传递。它在心脑血管,神经系统和免疫系统有很广泛的功能。已经证明内皮细胞、神经元和神经纤维末稍(非肾上腺和非胆碱神经纤维,NANC)以及神经胶质细胞内均有NO合酶(NOS)。现已克隆出3种重要的NOS(nNOS,eNOS和iNOS),在脑和内皮细胞中的NOS很相似,差别在于脑内的nNOS存在于细胞浆内,内皮的eNOS则与细胞膜相结合。从功能上NOS可分为原生型NOS(constitutive NOS,cNOS)和诱导型NOS(inducible NOS,iNOS)两类,前者包括nNOS和eNOS。
NO在脑循环和脑损伤过程中起重要作用。(1) 对脑循环作用:NO是很强的血管扩张剂,抑制血小板聚集和白细胞粘连。在生理状态下,由内皮、神经元和胶质细胞生成的NO作用于平滑肌细胞内的乌苷酸环化酶,生成cGMP,引起脑血管扩张,脑血流(CBF)增加。生成的NO还可通过负反馈限制NO的生成。持续的NO生成,使脑血管保持正常张力。在病理条件下,如在脑缺血时,脑缺血后2~3 min,NO迅速增加,6 min达高峰,60 min降至正常水平,若继续缺血,NO逐惭下降,当再灌时NO又增加。因此在缺血超早期,内皮产生NO超过神经元产生的有毒性的NO,能增加侧枝循环,阻止血小板和白细胞对微血管的阻塞,改善脑循环,这是其有利的一面。(2) 对脑损伤作用:NO有利和弊双重作用,其有利一面,除对改善CBF外,还能阻滞NMDA受体,这是由于NO氧化产生亚硝基离子,对NMDA受体亚硝基化,从而减轻兴奋毒对神经细胞的损伤。其有害的一面是在脑缺血数小时后,NO对血管不再有保作护用,而变为有害的了。在脑缺血超过6 h,iNOS在脑缺血后的炎症细胞部位表达,产生大量NO,引起迟发性损伤,过量NO对细胞有不利的影响,如NO与O()/(.)2作用能生成过氧化硝基产物(peroxynitrite),发挥细胞毒作用。NO还可引起能量耗竭,产生DNA损伤,抑制DNA合成,而且触发细胞凋亡。因此NO在脑缺血中的利和弊取决于脑缺血后的不同时间段和NO是由那种细胞所产生的。
, http://www.100md.com
过去对NOS抑制剂和NO生成剂对脑缺血的作用有矛盾报道。最近发现,NOS抑制剂如精氨酸类似物L-NMMA(L-甲基精氨酸),L-NNA(NG-硝基-L-精氨酸)和L-NAME(NG-硝基-L-精氨酸甲酯)等在低剂量或者治疗给药,使不抑制早期NO,则对脑缺血有保护作用,如用预防给药或大剂量治疗给药,由于抑制了早期NO则无保护作用,表现为脑梗塞体积增加。说明缺血早期NO增加是有利的因素,如被抑制,则对脑缺血不利。NOS的底物L-精氨酸能增加rCBF,降低梗塞体积。NO供体如硝普钠等也能降低脑梗塞体积。对NO供体和NOS抑制剂都能产生相同的脑保护作用则很难解释。最近有人用缺乏nNOS表达(用基因敲除技术)的突变小鼠或用选择性nNOS抑制剂7-NI进行实验,均能降低大脑中动脉阻断(MCAO)动物的梗塞体积。说明nNOS产生的NO在脑损伤中是不利的因素。相反,用缺乏eNOS表达的突变小鼠则能增加梗塞体积,说明内皮细胞产生的NO具保护作用。由此可见,有关影响NO药物的治疗前景,取决于是否能找到一个对NO产生过程有选择性作用的药物。如能选择性增加缺血早期NO生成或选择性抑制晚期NO生成的药物,或选择性抑制nNOS和iNOS作用的药物。
, http://www.100md.com
细胞因子和炎症反应[11,12] 脑缺血后白细胞参与微血管功能紊乱是继发性脑损伤的重要原因。在缺血脑区的微血管中血小板释放炎性介质PAF,它激活了多形核白细胞(PMNLs),PMNLs与内皮细胞粘连,促使内皮细胞皱缩,破裂,凝集坏死,使血脑屏障(BBB)破坏。粘附在内皮细胞上的PMNLs在趋化因子吸引下,通过内皮细胞层进入损伤脑区。产生急性炎性反应。由于BBB破坏,损伤脑区出现渗出液,发生水肿,坏死或有疤痕组织形成。在损伤脑区神经元释放细胞因子如TNF,IL-1和IL-6等,激活了巨噬细胞,它吞噬坏死神经元碎片,使轴突变性,出现类似外周组织的痊愈过程,如星形胶质细胞增生等现象。在脑缺血后一些具有免疫活性的细胞如内皮细胞,PMNLs和巨噬细胞,小神经胶质细胞和星形胶质细胞均能产生细胞因子和粘连分子。而这些细胞因子反过来又能激活上述细胞,这种正反馈作用,增加了内皮损伤,加强炎性反应。由于缺血脑区白细胞浸润结果,免疫细胞如内皮细胞和巨噬细胞释放化学趋化因子(chemokine),其作用是特异性地吸引单核细胞进入缺血脑区。总之,激活的吞噬细胞如小神经胶质细胞,游走的中性白细胞,血源性转换形成的巨噬细胞,免疫活性的淋巴细胞都可杀死脑细胞。从不同脑缺血动物模型和人脑卒中的缺血脑区均发现有免疫细胞如中性白血球和巨噬细胞的浸润。受刺激的脑细胞通过一种复杂的细胞因子网络(cytokine network)进行细胞间的信息传递,以调节每个细胞的功能状态。由此可见,在脑缺血继发性损伤中细胞因子,粘连分子所起的重要作用。细胞因子犹似细胞间的生理信使,它作为各种不同的神经病理调质,参与神经元的溃变与修复。从脑缺血后细胞因子,粘连分子和chemokine mRNA表达的时相来看,细胞因子和粘连分子的表达一般在大脑中动脉阻断后1~6 h检出,12 h达峰值,而chemokine mRNA的表达时相较细胞因子晚,一般在24~48 h达峰,推测chemokine mRNA的表达可能是由于细胞因子作用的结果。在脑卒中发作1小时后病人血清中的IL-6明显升高,10时达峰值,持续3日,第7日后降至正常,而未检测到IL-1β,TNFα和TNF-R。另有人发现卒中病人CSF中IL-6和IL-1β水平比血清中高。脑外伤时IL-1和IL-6在脑室液中明显升高。这表明动物实验与临床卒中病人有平行相关。
, 百拇医药
初步实验表明,用阻滞细胞因子的释放及阻滞其功能的方法,发现抗粘连分子抗体,失活循环中的白细胞,免疫抑制剂和PAF拮抗剂均能降低脑梗塞体积,也说明细胞因子在脑卒中的作用。
综上所述,脑缺血损伤的病理生理包含多个环节和多种因素的改变。假如能找到一个药物,能阻断多种不利环节和因素,则对缺血性脑卒中的治疗是极为有利的。
2 治疗急性脑缺血药物
溶栓药物 组织型纤溶酶原激活剂(t-PA)是一种丝氨酸蛋白酶,为内源性溶栓剂,它是在内皮细胞中产生。现已用DNA重组技术生产,称为rt-PA,是第2代溶栓药。它的分子是由527个氨基酸组成的多肽单链,在化学上与人的内源性t-PA相似。其作用机制与链激酶streptokinase(SK)和尿激酶urokinase(UK)相似,是将纤溶酶原转变为纤溶酶,使纤维蛋白裂解,而达到溶栓目的。在纤维蛋白存在下,它激活纤溶酶原的活性大大增加。它的优点是对循环中纤溶酶原的激活作用较SK弱,因此有可能减少出血合并症。使用时要选择适宜病例和治疗时间,溶栓的治疗窗范围一般在缺血性脑卒中症状发作后3~6 h内,越早疗效更佳。在颈动脉支配区因动脉闭塞出现症状后6~8 h内静脉滴注单或双链rt-PA,血管部分或完全再通率达34%~59%,而颅内出血率也较高(29%~53%)。最近有2项用重组组织型纤溶酶原激活剂(rt-PA,alteplase)治疗的临床试验报告。例如NINDS研究组[13]报道在卒中发作后3 h内静脉注射rt-PA收到较好效果,而卒中发作后36 h内颅内出血率较高(rt-PA:6.4%;安慰剂:0.6%),但能改善3个月后临床效果。Hacke[14]等报道一个多中心(14个欧洲国家,75个医院),随机、双盲,安慰剂对照试验,对患有中、重度神经功能缺损,CT检查无早期梗塞体症病人(n=620),在脑卒中发作后3或6 h内用rt-PA治疗,3个月后总结,rt-PA对急性脑卒中有较好的效果,但由于颅内出血率过高,不宜推荐用作未经选择的急性脑缺血病人的治疗药物。最近报道用动脉内溶栓治疗,再通率达(60~70)%,优于静脉内溶栓治疗。有3个用SK的临床试验报告,均因颅内出血和死亡率过高而中途中止试验。另有蛇毒制剂如东菱克栓酶(batroxobin,TOBISHI,DF-521)能分解纤维蛋白原,以致能溶解血栓,降低血液粘稠度,改善微循环,己在临床应用。虽然这些资料能鼓舞人们进一步试验,但这只能在有条件的医院和有经验的医师指导下才能进行。对近期有外伤,外科手术,胃肠道出血史者禁用。
, 百拇医药
钙拮抗剂尼莫地平(nimodipine,Nim) 属二氢吡啶类化合物,为第II类钙通道阻滞药。能减轻缺血性脑损伤,发现对神经元有直接保护作用。有关其药理作用和临床研究已有大量报道,很多研究表明它是一个有希望的治疗中枢神经系统的药物。其药理作用:(1) 能增加正常和脑缺血动物局部脑血流(rCBF),无窃血现象,一般伴有不同程度的血压下降。(2) 对蛛冈膜下腔出血(SAH)引起血管痉挛导致的CBF降低有改善作用。(3) 对局部和全脑脑缺血动物有预防和治疗作用。尼莫地平(Nim)的用途[15~18]:(1) 主要适应征是蛛冈膜下腔出血(SAH),能明显降低SAH发作后脑缺血和脑梗塞发生率,改善3个月的疗效。(2) 早期报道Nim能降低急性脑卒中病死率,改善神经功能。而近年来已有10余个多中心、随机、双盲和安慰剂对照临床试验。发现Nim治疗急性缺血性脑卒中的大部分临床试验看不到治疗效果。有一个较大规模临床试验,即美国Nim研究组报道1064例急性脑缺血病人,于发作后48 h内进行Nim治疗(120 mg.d-1),在治疗第1个月和第3个月Nim组的死亡率反较安慰剂组的高,但随诊一年后疗效与安慰剂组的相似。Nim治疗急性缺血性脑卒中的大部分临床试验看不到治疗效果,这是否与口服给药有关。改用注射剂后,有二个多中心(美国和欧洲)研究了Nim(iv)对急性缺血性脑卒中的效果。由于考虑安全问题,均于中途终止试验。说明Nim(po或iv)对缺血性脑卒中治疗效果并不肯定。
, http://www.100md.com
其他药物 这些药物尚在实验室研究阶段或在临床试验阶段。如(1) EAA拮抗剂:MK801为非竞争性NMDA受体拮抗剂,能明显降低脑缺血引起的神经元损伤,但由于有较严重副作用,限制了它的使用。此后,人们的注意力转为对非NMDA受体拮抗剂的研究,如AMPA受体拮抗剂DNQX,CNQX和NBQX,此外如GV150526(甘氨酸位点拮抗剂),selfotel和aptiganel等,但仍有较大副作用,它们中有的正在临床试用;(2) 自由基清除剂:据报道21位氨基类固醇化合物U74006F(商品名Tirilazad)具有强的抗实验性脑缺血作用,能降低MCAO大鼠脑梗塞体积,抑制自由基,阻断AA释放等。现在Tirilazad正进行III期临床试验,迄今尚未得出结论。现在对抗粘连分子及免疫抑制剂的研究也予以关注。
3 预防脑卒中的药物抗血小板药[19]
由于脑卒中发病率和死亡率均高,治疗方法虽在不断改进,而幸存者仍留下严重后遗症,生活不能自理,经济上耗费大。因此现在的谋略是希望能确定脑卒中高危人群,加以预防治疗,而主要手段是用抗血小板药物治疗,以降低血栓性脑卒中的危险因子,从而降低脑卒中的复发。
, http://www.100md.com
阿司匹林(aspirin) 是最为广泛应用的抗血小板药物,作用机制主要是对环氧合酶不可逆地乙酰化,使血小板环氧酶活性受到抑制。降低TXA2的生成。大剂量阿司匹林(ASA)也能抑制血管壁内皮细胞的环氧酶活性,使PGI2合成减少。因此适宜剂量可抑制实验性血栓形成,而大剂量ASA反而促使血栓生成。临床医生主张采用低剂量ASA预防脑卒中。最近发现ASA能抑制TNF剌激NF-kB的动员以及抑制TNF对VCAM-1(血管粘连分子-1)和E-Selectin(内皮细胞—选择蛋白)的诱导,进而抑制单核细胞与激活的内皮细胞的粘连,从而减轻内皮损伤,这一发现可能提供了另一种ASA的治疗作用机制。
有10余个多中心,随机,双盲安慰剂对照临床试验,均证明ASA对脑卒中二级预防有效[20,21],ASA大剂量(900~1300 mg.kg-1)和小剂量的效果相似。近年来大家已注意到了用小剂量ASA(30,50或75 mg.d-1)进行脑卒中的二级预防收到较好效果。
, http://www.100md.com
最近CAST(Chinese Acute Stroke Trial) Collaborative Group[22](中国急性脑卒中试验协作组)用多中心、随机、安慰剂对照试验得到了与IST(International Stroke Trial)[23]相似的结果:发现急性缺血性脑卒中病人发病后48 h内口服ASA(160 mg.d-1,连服4周,n=10554)治疗,与安慰剂组(n=10552)比较,死亡率降低14%,卒中复发率低。用死亡率和非致命脑卒中发生率为指标,降低12%。说明ASA不仅预防有效,而且治疗早期急性脑卒中有效。
ASA主要出现肠胄道刺激不良反应。少数病人由于血小板功能抑制和出血时间延长而导致脑内或胃肠道出血。
双嘧达莫(dipyridamole) 又名潘生丁(persantine),其抑制血小板功能主要是通过对血小板磷酸二脂酶抑制而升高cAMP,以致降低血小板粘连和聚集。并且它阻止红细胞及血管内皮细胞对腺苷(Ade)的摄取和代谢,以致增加了Ade在血浆中浓度,激活了腺苷环化酶,增加cAMP而抑制血小板聚集。双嘧达莫(DP)对脑卒中二级预防是否有效一直未加肯定。最近,肯定DP对脑卒中二级预防有效的一个临床试验是来自ESPS2的报告[21]。有13个欧洲国家参与试验,他们采用安慰剂对照和双盲试验,随机分成4个组:低剂量ASA 50 mg.d-1 (n=1649)组,DP缓释剂型组400 mg.d-1 (n=1654),ASA合并服用DP组(n=1650)以及安慰剂组(n=1649)。随诊2年,以脑卒中复发和死亡率为指标。与安慰剂组比较,发现卒中发病率分别降低18.1%(ASA),16.3%(DP)和37%(ASA+DP)。脑卒中发作和死亡率分别降低13.2%,15.4%和24.4%。从这一规模较大的试验结果可以看出DP是有效的,而且与ASA合并用药其疗效有增强作用。
, 百拇医药
噻氯匹定(ticlopidine) 是一新型的抗血小板药,它抑制血小板功能是通过抑制ADP及多种激动剂引起的血小板聚集。可能的机制是选择性干扰ADP诱发的纤维蛋白原与糖蛋白IIb/IIIa复合物的结合。对磷脂酶A活性及对AA代谢物均无影响。对cAMP-AC系统也无直接作用。一般口服后24~48 h开始起效,3~5 d作用最强,停药后药效尚能维持72 h。
有数个多中心、随机、双盲和安慰剂对照试验,噻氯匹定均能有效地预防TIA复发[24,25]。Bellavance报道一个多中心、双盲、随机临床试验表明,噻氯匹定剂量为250 mg.d-1 (n=1529),ASA为650 mg.d-1 (n=1540),随诊5.8年,噻氯匹定和ASA均明显降低脑卒中危象,但前者疗效略好。但由于价贵及较严重的中性粒细胞减少的副作用,因此主张适用于不能耐受ASA的病人,或在ASA治疗期间有缺血性脑卒中发作的患者。
, 百拇医药
根据上述抗血小板药的临床试验资料,凡患者有近期TIA史,或有轻度完全性脑卒中而CT未证实有脑出血病人均可用抗血小板药物进行二级预防。Matchar等[26]综述了自1977年来发表的应用抗栓药如ASA,DP,噻氯匹定或硫氧唑酮预防脑血管病的概况。选择随机和安慰剂对比临床试验材料为依据,得出以下结论:认为(1) TIA或轻度脑卒中患者ASA为首选药物;(2) 对ASA无反应或耐受者,而且在治疗期间发生脑卒中者可用噻氯匹定治疗;(3) 在接受抗凝治疗有出血危险因子者可选用ASA。
4 治疗脑血管病恢复期和后遗症的药物
这类药物的作用性质大多数为脑代谢和脑循环改善剂。有些药物有较好的药效学实验基础,而有的则较为粗糙,在我国临床上应用较多的药物有:
银杏叶提取物(Ginkgo biloba extracts,GBE)[27] 银杏是一种古老的植物,其果实为传统中药。自60年代始,西方国家从叶中提取并分离其成分。有药理活性的成份有两种:一类是黄酮类似物(flavonoids)另一类是银杏苦内酯(即ginkgolides,或terpenes),从中已分离出银杏苦苷A,B,C,J和M等单体。其中以银杏苦苷B(BN 52021)的作用最强,A(BN 52020)次之,余较弱。银杏提取物中主要含黄酮苷和银杏苦内酯。
, 百拇医药
其药理作用为:(1) 黄酮类似物有清除自由基,抑制脂质过氧化的作用。能扩张血管,增加脑血流量。(2) 银杏苦内酯则对PAF有特异性抑制作用,因此能对抗血小板聚集和防止血栓形成作用。(3) BN 52021能抑制脑组织内水、Na+蓄积和K+的丢失,减轻脑水肿的形成。(4) GBE对实验性局部脑缺血有一定保护作用,降低多发性脑梗塞大鼠BBB的损伤,使减轻脑水肿;能改善缺血脑ATP合成;对实验性引起兔SAH的血管痉挛呈明显的抑制作用。GBE的主要适应证为脑功能不全。
艾地苯醌(idebenone) 药理作用:(1) 改善脑功能障碍,(2) 改善脑能量代谢,(3) 对线粒体肿胀和脂质过氧化有抑制作用。可用于改善脑代谢和精神症状。
其他 丹参注射剂,丹参对实验性脑缺血研究较少,据报道丹参有抗体外血栓形成和抗血小板聚集功能,对收缩状态的微血管有显著的扩张作用,可改善微循环障碍,改善细胞缺血、缺氧所致的代谢障碍。临床试验认为有一定效果;长春西丁(vinpocetine,商品名为卡兰)为脑循环和脑代谢改善剂,对脑缺血有保护作用。用于治疗脑卒中后遗症,脑动脉硬化症。但尚缺乏严格的多中心,随机,双盲,安慰剂对照试验的检验。另有氢化麦角碱、川芎嗪、二苯美伦(befemelane)、环扁桃酯(cyclandelate,商品名为抗栓丸)、胞二磷胆碱(citilcholine)、已酮可可碱(pentoxifylline)和中药神农丸等的临床适应证较相似,多数适用于脑卒中恢复期和后遗症。
, 百拇医药
5 本实验室研究开发的新药
作者实验室从整体动物、细胞和分子生物学等不同水平,对抗脑缺血新药丁苯酞进行了较广泛而深入的研究。丁苯酞(dl-butylphthalide,NBP)是从芹菜籽中提取出来的左旋体,后经人工合成为消旋体。经研究发现NBP抗脑缺血的药效学作用如下:(1) NBP(150~200 mg.kg-1 sc)具有改善小鼠全脑缺血脑能量代谢,即能逆转缺血时ATP和PCr的降低和LA的升高,使其恢复和接近正常水平[28]。(2) NBP(10~20 mg.kg-1 ip)能增加局部脑缺血大鼠(MCAO)缺血区的局部脑血流(rCBF)[29]。(3) NBP(10~20 mg.kg-1 ip或40~160 mg.kg-1 po)能缩小MCAO大鼠脑梗塞面积和改善神经功能缺失[30]。(4) NBP(80~240 mg.kg-1 po)能减轻MCAO大鼠脑水肿[31],(5) NBP(30~100 mg.kg-1 po)能改善MCAO大鼠记忆功能障碍[32]。(6) NBP(100 mg.kg-1 po)对脑卒中易感型自发性高血压大鼠有预防和治疗作用[33]。(7) NBP(1~100 μmol.L-1)对多种原因如高钾、兴奋毒和低氧低糖引起的培养胎大鼠神经细胞损伤有直接保护作用[34]。
, http://www.100md.com
对NBP作用机制研究表明:(1) NBP(5~10 mg.kg-1 ip)对t-MCAO(暂时性MCAO)大鼠脑线粒体呼吸链复合酶IV活性抑制有明显逆转作用[35],提示改善缺血脑能量代谢(增加ATP,PCr,降低LA)是通过这一作用而发挥的;(2) NBP有降低大鼠尾动脉环细胞内钙的作用,而对外钙无影响[36],对神经细胞内钙也有明显降低作用(待发表资料);(3) 发现d-NBP可激活大鼠脑突触体胞液cNOS活性,增加NO和cGMP,对iNOS无作用,而l-NBP则能抑制cNOS和iNOS。随后的实验表明,蛛网膜下腔出血引起的rCBF降低,d-NBP能改善rCBF,而l-NBP则无影响。由此推测,NBP可能是通过激活cNOS而增加rCBF(待发表资料);(4) NBP(20~40 mg.kg-1 ip)能抑制全脑缺血(4血管阻断)大鼠纹状体细胞外液ATP降解代谢产物次黄嘌呤(Hyp)和黄嘌呤(Xan)的含量升高[37],提示O()/(.)2底物生成减少,降低了的过量产生;(5) NBP(10~20 mg.kg-1 ip)能明显抑制t-MCAO大鼠脑皮层缺血再灌期AA含量的增加(待发表资料);(6) NBP能明显抑制局部脑缺血大鼠缺血再灌期脑MDA的产生,体外实验表明NBP能抑制黄嘌呤和黄嘌呤氧化酶所生成的(待发表资料);(7) NBP能降低有害刺激对胎大鼠培养神经细胞谷氨酸(Glu)释放的增加;(8) 能增加局部脑缺血大鼠缺血再灌期PGI2/TXA2的比值[38];(9) 用原位杂交法和Nothern印迹杂交法证明NBP能明显抑制局部脑缺血大鼠缺血再灌后12和24 h的hsp70mRNA的表达[39]。由上述结果可见,NBP作用于多个病理环节:如改善线粒体功能和脑微循环,降低Glu的释放,抑制[Ca2+]i和AA,抑制和MDA,特别是前两者是它发挥抗脑缺血的主要作用。由于局部脑缺血的脑损伤在始初和迟发性损伤过程中是一多因素参与的过程,如在治疗上只解决复杂过程中的一个方面,恐怕不会收到好的效果。因此在治疗上去阻断脑缺血病理过程的多个环节是很重要的。而NBP正是具有作用于多个病理环节的药物,这对其开发为抗脑缺血药物具有良好的前景。现在NBP正在进行II期临床试验。
, http://www.100md.com
6 结语和展望
急性缺血性脑卒中严重危害着人类健康,幸存者往往留下后遗症,影响病人生活质量,也增加家庭和社会负担。为此,探索其病理生理,研究药物干预极为重要。自80年代以来,很多学者除提出能量代谢、兴奋毒、氧化损伤、钙超载等学说外、最近也注意到脑缺血与细胞因子、炎症反应、粘连分子等密切相关。相应的药物也应运而出,如钙拮抗剂、兴奋性受体拮抗剂和自由基清除剂等,但能否治疗急性缺血性脑卒中仍不能完全肯定。临床上用溶栓药如t-PA等治疗缺血性脑卒中(在发作后6 h内用药)虽收到较好效果,但尚未解决出血率高的危险性。由于缺乏治疗药物,现在各国多用预防药如阿司匹林等进行二级预防,取得较好效果。近年来,各国学者对缺血性脑损伤的病理生理进行了深入探讨,企图寻找一个能阻断缺血性脑损伤的多个病理环节的新药,相信这一天将会到来。
关键词 脑缺血;一氧化氮;细胞因子;炎症反应;丁基苯酞
联系人 Tel:(010)63165173, Fax:(010)63017757,E-mail:fengyp@public.gb.com.cn
, 百拇医药
参考文献
1 李世绰.中国循环杂志,1993,8∶577
2 冯亦璞.陈维洲等主编.心血管药理学进展.北京:人民卫生出版社,1995.90
3 Siesjo BK, Katsura KI, Zhao Q, et al. J Neurotrauma, 1995,12∶943
4 MacManus JP, Linnik MD. J Cereb Blood Flow Metab, 1997,17∶815
5 Beckman JS, Ye YZ, Chen J, et al. Advances in Neurology. Vol 71. In: Siesjo Bk, Wieloch T, ed. Philadelphia: Lippinocott-Raven Publihers, 1996.339
, 百拇医药
6 The RANTTAS Investigators. Stroke, 1996,27∶1453
7 Faraci FM, Brain JE. Stroke, 1994,25∶692
8 Siesjo Bk, Wieloch T, ed. Advances in Neurology. Vol 71. Philadelphia: Lippinocott-Raven Pubblihers, 1996.355
9 Samdani AF, Dawson TM, Dawson VL. Stroke, 1997,28∶1283
10 Iadecola C. Trends Neurosci, 1997,20∶132
11 Tomita M, Fukuuchi Y. Acta Neurochim, 1996,66(Suppl)∶32
, 百拇医药
12 Kim JS. J Neurosci, 1996,137∶67
13 The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. N Engl J Med, 1995,333∶1581
14 Hacke W, Kaste M, Fieschi C, et al. J Am Med Assoc, 1995,274∶1017
15 Pickard JD, Murray GD, Illingworth R, et al. Br Med J, 1989,298∶636
16 Harders A, Kakarieka A, Braakman R, et al. J Neurosurg, 1996,85∶82
, http://www.100md.com
17 The American Nimodipine Study Group. Stroke, 1992,23∶3
18 Kaste M, Fogelholm R, Erila T. Stroke, 1994,25∶1348
19 汪钟,郑植荃主编.现代血栓病学.北京:北京医科大学中国协和医科大学联合出版社,1997.508
20 The SALT Collaborative Group. Lancet, 1991,338∶1345
21 Diener HC, Cunha L, Forbes C, et al. J Neurol Sci, 1996,143∶1
22 CAST (Chinese Acute Stroke Trial) Collaborative group. Lancet, 1997,349∶1641
, http://www.100md.com
23 International Stroke Trial Collaborative Group. Lancet, 1997,349∶1569
24 Gent M, Blakely JA, Easton JD, et al. Lancet, 1989,1∶1215
25 Bellavance A. Stroke, 1993,24∶1452
26 Matchar DB, McCrory DC, Barnett HJ, et al. Ann Inter Med, 1994,121∶41
27 陈维洲,芮耀诚主编.脑血管病基础与临床研究.济南:山东科学技术出版社,1993∶153
28 冯亦璞,胡盾,张丽英.药学学报,1995,30∶741
, 百拇医药
29 Yan CH, Feng YP, Zhang JT. Acta Pharmacol Sin, 1998,19∶117
30 刘小光,冯亦璞.药学学报,1995,30∶896
31 Deng WB, Feng YP. Chin Med Sci J, 1997,12∶102
32 胡盾,张丽英,冯亦璞.中国药理学与毒理学杂志,1997,11∶14
33 张丽英,冯亦璞.药学学报,1996,31∶18
34 阎超华,张均田,冯亦璞.药学学报,1997,32∶340
35 熊杰,冯亦璞.药学学报,1999,34∶(印刷中)
, 百拇医药
36 刘小光,冯亦璞.中国药理学与毒理学杂志,1996,10∶113
37 胡盾,黄新祥,冯亦璞.药学学报,1996,31∶13
38 Chong ZZ, Feng YP. Acta Pharmacol Sin, 1997,18∶505
39 熊杰,冯亦璞.药学学报,1998,33∶401
PATHOPHYSIOLOGY OF ISCHEMIC STROKE
AND STATUS OF DRUG INTERVENTION
Feng Yipu(Feng YP)
(Institute of Materia Medica, Chinese Academy of Medical Sciences
and Peking Union Medical College, Beijing 100050, China)
KEY WORDS cerebral ischemia; nitric oxide; cytokine; inflammatory reaction; dl-butyphthalide
收稿日期: 1998-11-10, 百拇医药
单位:(中国医学科学院、中国协和医科大学药物研究所,北京 100050)
关键词:
药学学报990117.htm 目前脑血管病已成为我国3大病死原因之一[1]。对脑卒中病理生理的深入研究加深了对其预防和治疗的认识,并推动和加速对新型抗脑缺血药物的研制开发。
脑卒中大体可分成两大类:(1) 缺血性脑卒中,约占(70~80)%;(2) 出血性脑卒中,约占(20~30)%。这两类病变在治疗上完全不同。本文着重介绍缺血性脑卒中的病理生理和药物治疗。
1 脑缺血病理生理改变的因素[2]
虽然近年来,由于钙超载,毒性氧自由基和兴奋毒等学说的出现,为解释脑缺血的病理生理奠定了基础,一些治疗脑缺血的药物如钙通道阻滞药、自由基清除剂和兴奋性氨基酸(EAA)拮抗剂也应运而生,但迄今尚无较理想的治疗药物。因此,对脑缺血病理生理的深入研究仍是艰巨任务。Siesjo[3]认为全脑缺血时钙通过谷氨酸受体门控性和电压依赖性通道进入胞内。钙超载后触发了一系列影响再灌期神经元存活的反应,引起神经元存活后数小时乃至数日后继发性死亡。始初的损伤可能引起细胞膜钙处理能力的持续变化,导致线粒体持续、缓慢的钙超载。另一种可能是细胞内的信号转导通路持续紊乱而引起转录和翻译水平上的改变,及某些细胞(如产生应激蛋白的细胞,产生营养因子或对存活必需酶的细胞)的丢失。而在这一变化过程中,并未见微血管或线粒体功能障碍。与此相反,局部脑缺血虽然也会引起钙超载而触发一系列反应,但对脑细胞死亡的作用是次要的,而起主要作用的是在继发性损伤中产生的调质所引起的反应。这些调质可能是花生四烯酸(AA)代谢物,NO,自由基或其它活性代谢物。在缺血期或缺血1~3 h后的再灌期,这些调质可诱导内皮细胞和多形核白细胞(PMNLs)上粘连分子的表达或氧化关键蛋白质,还可触发炎性细胞因子如白介素(IL)-1,-6,-8,γ-干扰素,或肿瘤坏死因子(TNF)的合成和释放,引起缺血阴影区微血管阻塞。此外可产生由于持续缺血所引起的线粒体功能衰竭。因此局部脑缺血时微循环和线粒体功能衰竭是引起缺血继发性损伤的主要原因。近年来,在对继发性脑损伤中细胞因子和炎症反应所起的作用也已受到重视,以下就脑缺血后病理生理改变的多种因素,特别是对近年来一些新的看法作重点介绍。
, 百拇医药
细胞凋亡[4] 是指细胞按自身程序主动死亡的过程,而细胞坏死则是被动死亡过程。细胞凋亡在形态学和生化特徵上表现为细胞皱缩,细胞核染色质浓缩,DNA片断化,而细胞的膜结构和细胞器仍完整。研究发现,脑缺血后迟发性神经元损伤过程出现细胞凋亡,有人认为迟发性神经元损伤主要是由细胞凋亡所致,但对此仍有不同看法。在缺血缺氧时有几种因素调节细胞的生、死命运。一般认为,在抑制细胞凋亡过程有信号转导,如神经营养因子和应激蛋白等,它们分别激活抑制凋亡的基因如bcl2/bclXL等的表达,使细胞凋亡受到抑制,从而改善脑损伤;而在促进细胞凋亡过程中出现bax/bad和p53等基因的表达,激活某些蛋白水解酶,使修复DNA的酶如PARP(poly(ADP-ribose)polymerase)和DNA-PKcs(DNA-dependent protein kinase)等降解,最后导至细胞凋亡。由此可见,探讨凋亡与相关基因或相关因素(线粒体功能受损如ATP合成受阻、膜电位失衡、氧化损伤以及蛋白水解酶的激活等)之间的关系是很重要的,这为寻找治疗脑损伤药物提供线索。
, 百拇医药
自由基 脑缺血再灌期,当细胞内Ca2+浓度升高时,胞内依赖于Ca2+的蛋白水解酶被激活,黄嘌呤脱氢酶转变为黄嘌呤氧化酶(XO)。与此同时,ATP降解代谢物如次黄嘌呤(HPY)和黄嘌呤(Xan)增加。在XO作用下,产生大量毒性氧自由基。此外,花生四烯酸(AA)经环氧酶代谢途径产生
,在铁蛋白存在下,转变为.OH,攻击膜结构和DNA,使细胞组织结构和功能均遭到破坏。此外,NO途径所产生的氧自由基也起重要作用[5]。在生理情况下,NO与的反应速度比超氧物歧化酶(SOD)清除的速度快3倍,但由于体内NO浓度(10~100 nmol.L-1)比体内SOD的浓度大约低100倍,此时产生的NO很难与SOD竞争,而主要作为信使传导分子行使其功能。在病理情况下如脑缺血/再灌时,细胞内NO的浓度显著升高,以至于达到能与SOD竞争超氧化物的水平,因为NO含有一个未配对电子,有存在时,迅速与其反应生成硝基过氧化物(ONOO—),后者质子化后进一步生成过氧亚硝酸(ONOOH),并在酸性环境中进一步分解为.OH和NO2。过去认为,.OH是NO引起脑损伤的最重要的氧自由基,现在则认为硝基过氧化物的直接氧化作用的毒性远大于.OH的毒性,因为硝基过氧化物不仅是一种很强的氧化剂,更重要的是它对反应有很高的选择性,如过氧化亚硝基能够直接氧化脂质、DNA以及蛋白质的巯基、锌/硫中心及铁/硫中心等,上述反应的速率大约是过氧化亚硝酸分解产生.OH速率的1 000倍。由此可见,硝基过氧化物可能是NO途径中产生的一种非常重要的氧自由基,以致严重损伤神经元。有关从抗氧自由基的途径去研究抗脑缺血药物也受到重视。据报道,维生素E和21位氨基类固醇化合物U74006F(商品名tirilazad)具有强的抗实验性脑缺血作用。对U74006F进行了多中心、随机、双盲和安慰剂对照临床试验,于脑卒中后4.3 h给药(6 mg.kg.d-1×3),未见神经功能改善[6]。, http://www.100md.com
一氧化氮[7~10] 它是一个能扩散的有高度化学活性的气体分子,半衰期短,可在细胞间自由地传递信使。而与经典的神经介质不同,它能在神经元之间或神经元与胶质细胞之间进行顺向和逆向的信息传递。它在心脑血管,神经系统和免疫系统有很广泛的功能。已经证明内皮细胞、神经元和神经纤维末稍(非肾上腺和非胆碱神经纤维,NANC)以及神经胶质细胞内均有NO合酶(NOS)。现已克隆出3种重要的NOS(nNOS,eNOS和iNOS),在脑和内皮细胞中的NOS很相似,差别在于脑内的nNOS存在于细胞浆内,内皮的eNOS则与细胞膜相结合。从功能上NOS可分为原生型NOS(constitutive NOS,cNOS)和诱导型NOS(inducible NOS,iNOS)两类,前者包括nNOS和eNOS。
NO在脑循环和脑损伤过程中起重要作用。(1) 对脑循环作用:NO是很强的血管扩张剂,抑制血小板聚集和白细胞粘连。在生理状态下,由内皮、神经元和胶质细胞生成的NO作用于平滑肌细胞内的乌苷酸环化酶,生成cGMP,引起脑血管扩张,脑血流(CBF)增加。生成的NO还可通过负反馈限制NO的生成。持续的NO生成,使脑血管保持正常张力。在病理条件下,如在脑缺血时,脑缺血后2~3 min,NO迅速增加,6 min达高峰,60 min降至正常水平,若继续缺血,NO逐惭下降,当再灌时NO又增加。因此在缺血超早期,内皮产生NO超过神经元产生的有毒性的NO,能增加侧枝循环,阻止血小板和白细胞对微血管的阻塞,改善脑循环,这是其有利的一面。(2) 对脑损伤作用:NO有利和弊双重作用,其有利一面,除对改善CBF外,还能阻滞NMDA受体,这是由于NO氧化产生亚硝基离子,对NMDA受体亚硝基化,从而减轻兴奋毒对神经细胞的损伤。其有害的一面是在脑缺血数小时后,NO对血管不再有保作护用,而变为有害的了。在脑缺血超过6 h,iNOS在脑缺血后的炎症细胞部位表达,产生大量NO,引起迟发性损伤,过量NO对细胞有不利的影响,如NO与O()/(.)2作用能生成过氧化硝基产物(peroxynitrite),发挥细胞毒作用。NO还可引起能量耗竭,产生DNA损伤,抑制DNA合成,而且触发细胞凋亡。因此NO在脑缺血中的利和弊取决于脑缺血后的不同时间段和NO是由那种细胞所产生的。
, http://www.100md.com
过去对NOS抑制剂和NO生成剂对脑缺血的作用有矛盾报道。最近发现,NOS抑制剂如精氨酸类似物L-NMMA(L-甲基精氨酸),L-NNA(NG-硝基-L-精氨酸)和L-NAME(NG-硝基-L-精氨酸甲酯)等在低剂量或者治疗给药,使不抑制早期NO,则对脑缺血有保护作用,如用预防给药或大剂量治疗给药,由于抑制了早期NO则无保护作用,表现为脑梗塞体积增加。说明缺血早期NO增加是有利的因素,如被抑制,则对脑缺血不利。NOS的底物L-精氨酸能增加rCBF,降低梗塞体积。NO供体如硝普钠等也能降低脑梗塞体积。对NO供体和NOS抑制剂都能产生相同的脑保护作用则很难解释。最近有人用缺乏nNOS表达(用基因敲除技术)的突变小鼠或用选择性nNOS抑制剂7-NI进行实验,均能降低大脑中动脉阻断(MCAO)动物的梗塞体积。说明nNOS产生的NO在脑损伤中是不利的因素。相反,用缺乏eNOS表达的突变小鼠则能增加梗塞体积,说明内皮细胞产生的NO具保护作用。由此可见,有关影响NO药物的治疗前景,取决于是否能找到一个对NO产生过程有选择性作用的药物。如能选择性增加缺血早期NO生成或选择性抑制晚期NO生成的药物,或选择性抑制nNOS和iNOS作用的药物。
, http://www.100md.com
细胞因子和炎症反应[11,12] 脑缺血后白细胞参与微血管功能紊乱是继发性脑损伤的重要原因。在缺血脑区的微血管中血小板释放炎性介质PAF,它激活了多形核白细胞(PMNLs),PMNLs与内皮细胞粘连,促使内皮细胞皱缩,破裂,凝集坏死,使血脑屏障(BBB)破坏。粘附在内皮细胞上的PMNLs在趋化因子吸引下,通过内皮细胞层进入损伤脑区。产生急性炎性反应。由于BBB破坏,损伤脑区出现渗出液,发生水肿,坏死或有疤痕组织形成。在损伤脑区神经元释放细胞因子如TNF,IL-1和IL-6等,激活了巨噬细胞,它吞噬坏死神经元碎片,使轴突变性,出现类似外周组织的痊愈过程,如星形胶质细胞增生等现象。在脑缺血后一些具有免疫活性的细胞如内皮细胞,PMNLs和巨噬细胞,小神经胶质细胞和星形胶质细胞均能产生细胞因子和粘连分子。而这些细胞因子反过来又能激活上述细胞,这种正反馈作用,增加了内皮损伤,加强炎性反应。由于缺血脑区白细胞浸润结果,免疫细胞如内皮细胞和巨噬细胞释放化学趋化因子(chemokine),其作用是特异性地吸引单核细胞进入缺血脑区。总之,激活的吞噬细胞如小神经胶质细胞,游走的中性白细胞,血源性转换形成的巨噬细胞,免疫活性的淋巴细胞都可杀死脑细胞。从不同脑缺血动物模型和人脑卒中的缺血脑区均发现有免疫细胞如中性白血球和巨噬细胞的浸润。受刺激的脑细胞通过一种复杂的细胞因子网络(cytokine network)进行细胞间的信息传递,以调节每个细胞的功能状态。由此可见,在脑缺血继发性损伤中细胞因子,粘连分子所起的重要作用。细胞因子犹似细胞间的生理信使,它作为各种不同的神经病理调质,参与神经元的溃变与修复。从脑缺血后细胞因子,粘连分子和chemokine mRNA表达的时相来看,细胞因子和粘连分子的表达一般在大脑中动脉阻断后1~6 h检出,12 h达峰值,而chemokine mRNA的表达时相较细胞因子晚,一般在24~48 h达峰,推测chemokine mRNA的表达可能是由于细胞因子作用的结果。在脑卒中发作1小时后病人血清中的IL-6明显升高,10时达峰值,持续3日,第7日后降至正常,而未检测到IL-1β,TNFα和TNF-R。另有人发现卒中病人CSF中IL-6和IL-1β水平比血清中高。脑外伤时IL-1和IL-6在脑室液中明显升高。这表明动物实验与临床卒中病人有平行相关。
, 百拇医药
初步实验表明,用阻滞细胞因子的释放及阻滞其功能的方法,发现抗粘连分子抗体,失活循环中的白细胞,免疫抑制剂和PAF拮抗剂均能降低脑梗塞体积,也说明细胞因子在脑卒中的作用。
综上所述,脑缺血损伤的病理生理包含多个环节和多种因素的改变。假如能找到一个药物,能阻断多种不利环节和因素,则对缺血性脑卒中的治疗是极为有利的。
2 治疗急性脑缺血药物
溶栓药物 组织型纤溶酶原激活剂(t-PA)是一种丝氨酸蛋白酶,为内源性溶栓剂,它是在内皮细胞中产生。现已用DNA重组技术生产,称为rt-PA,是第2代溶栓药。它的分子是由527个氨基酸组成的多肽单链,在化学上与人的内源性t-PA相似。其作用机制与链激酶streptokinase(SK)和尿激酶urokinase(UK)相似,是将纤溶酶原转变为纤溶酶,使纤维蛋白裂解,而达到溶栓目的。在纤维蛋白存在下,它激活纤溶酶原的活性大大增加。它的优点是对循环中纤溶酶原的激活作用较SK弱,因此有可能减少出血合并症。使用时要选择适宜病例和治疗时间,溶栓的治疗窗范围一般在缺血性脑卒中症状发作后3~6 h内,越早疗效更佳。在颈动脉支配区因动脉闭塞出现症状后6~8 h内静脉滴注单或双链rt-PA,血管部分或完全再通率达34%~59%,而颅内出血率也较高(29%~53%)。最近有2项用重组组织型纤溶酶原激活剂(rt-PA,alteplase)治疗的临床试验报告。例如NINDS研究组[13]报道在卒中发作后3 h内静脉注射rt-PA收到较好效果,而卒中发作后36 h内颅内出血率较高(rt-PA:6.4%;安慰剂:0.6%),但能改善3个月后临床效果。Hacke[14]等报道一个多中心(14个欧洲国家,75个医院),随机、双盲,安慰剂对照试验,对患有中、重度神经功能缺损,CT检查无早期梗塞体症病人(n=620),在脑卒中发作后3或6 h内用rt-PA治疗,3个月后总结,rt-PA对急性脑卒中有较好的效果,但由于颅内出血率过高,不宜推荐用作未经选择的急性脑缺血病人的治疗药物。最近报道用动脉内溶栓治疗,再通率达(60~70)%,优于静脉内溶栓治疗。有3个用SK的临床试验报告,均因颅内出血和死亡率过高而中途中止试验。另有蛇毒制剂如东菱克栓酶(batroxobin,TOBISHI,DF-521)能分解纤维蛋白原,以致能溶解血栓,降低血液粘稠度,改善微循环,己在临床应用。虽然这些资料能鼓舞人们进一步试验,但这只能在有条件的医院和有经验的医师指导下才能进行。对近期有外伤,外科手术,胃肠道出血史者禁用。
, 百拇医药
钙拮抗剂尼莫地平(nimodipine,Nim) 属二氢吡啶类化合物,为第II类钙通道阻滞药。能减轻缺血性脑损伤,发现对神经元有直接保护作用。有关其药理作用和临床研究已有大量报道,很多研究表明它是一个有希望的治疗中枢神经系统的药物。其药理作用:(1) 能增加正常和脑缺血动物局部脑血流(rCBF),无窃血现象,一般伴有不同程度的血压下降。(2) 对蛛冈膜下腔出血(SAH)引起血管痉挛导致的CBF降低有改善作用。(3) 对局部和全脑脑缺血动物有预防和治疗作用。尼莫地平(Nim)的用途[15~18]:(1) 主要适应征是蛛冈膜下腔出血(SAH),能明显降低SAH发作后脑缺血和脑梗塞发生率,改善3个月的疗效。(2) 早期报道Nim能降低急性脑卒中病死率,改善神经功能。而近年来已有10余个多中心、随机、双盲和安慰剂对照临床试验。发现Nim治疗急性缺血性脑卒中的大部分临床试验看不到治疗效果。有一个较大规模临床试验,即美国Nim研究组报道1064例急性脑缺血病人,于发作后48 h内进行Nim治疗(120 mg.d-1),在治疗第1个月和第3个月Nim组的死亡率反较安慰剂组的高,但随诊一年后疗效与安慰剂组的相似。Nim治疗急性缺血性脑卒中的大部分临床试验看不到治疗效果,这是否与口服给药有关。改用注射剂后,有二个多中心(美国和欧洲)研究了Nim(iv)对急性缺血性脑卒中的效果。由于考虑安全问题,均于中途终止试验。说明Nim(po或iv)对缺血性脑卒中治疗效果并不肯定。
, http://www.100md.com
其他药物 这些药物尚在实验室研究阶段或在临床试验阶段。如(1) EAA拮抗剂:MK801为非竞争性NMDA受体拮抗剂,能明显降低脑缺血引起的神经元损伤,但由于有较严重副作用,限制了它的使用。此后,人们的注意力转为对非NMDA受体拮抗剂的研究,如AMPA受体拮抗剂DNQX,CNQX和NBQX,此外如GV150526(甘氨酸位点拮抗剂),selfotel和aptiganel等,但仍有较大副作用,它们中有的正在临床试用;(2) 自由基清除剂:据报道21位氨基类固醇化合物U74006F(商品名Tirilazad)具有强的抗实验性脑缺血作用,能降低MCAO大鼠脑梗塞体积,抑制自由基,阻断AA释放等。现在Tirilazad正进行III期临床试验,迄今尚未得出结论。现在对抗粘连分子及免疫抑制剂的研究也予以关注。
3 预防脑卒中的药物抗血小板药[19]
由于脑卒中发病率和死亡率均高,治疗方法虽在不断改进,而幸存者仍留下严重后遗症,生活不能自理,经济上耗费大。因此现在的谋略是希望能确定脑卒中高危人群,加以预防治疗,而主要手段是用抗血小板药物治疗,以降低血栓性脑卒中的危险因子,从而降低脑卒中的复发。
, http://www.100md.com
阿司匹林(aspirin) 是最为广泛应用的抗血小板药物,作用机制主要是对环氧合酶不可逆地乙酰化,使血小板环氧酶活性受到抑制。降低TXA2的生成。大剂量阿司匹林(ASA)也能抑制血管壁内皮细胞的环氧酶活性,使PGI2合成减少。因此适宜剂量可抑制实验性血栓形成,而大剂量ASA反而促使血栓生成。临床医生主张采用低剂量ASA预防脑卒中。最近发现ASA能抑制TNF剌激NF-kB的动员以及抑制TNF对VCAM-1(血管粘连分子-1)和E-Selectin(内皮细胞—选择蛋白)的诱导,进而抑制单核细胞与激活的内皮细胞的粘连,从而减轻内皮损伤,这一发现可能提供了另一种ASA的治疗作用机制。
有10余个多中心,随机,双盲安慰剂对照临床试验,均证明ASA对脑卒中二级预防有效[20,21],ASA大剂量(900~1300 mg.kg-1)和小剂量的效果相似。近年来大家已注意到了用小剂量ASA(30,50或75 mg.d-1)进行脑卒中的二级预防收到较好效果。
, http://www.100md.com
最近CAST(Chinese Acute Stroke Trial) Collaborative Group[22](中国急性脑卒中试验协作组)用多中心、随机、安慰剂对照试验得到了与IST(International Stroke Trial)[23]相似的结果:发现急性缺血性脑卒中病人发病后48 h内口服ASA(160 mg.d-1,连服4周,n=10554)治疗,与安慰剂组(n=10552)比较,死亡率降低14%,卒中复发率低。用死亡率和非致命脑卒中发生率为指标,降低12%。说明ASA不仅预防有效,而且治疗早期急性脑卒中有效。
ASA主要出现肠胄道刺激不良反应。少数病人由于血小板功能抑制和出血时间延长而导致脑内或胃肠道出血。
双嘧达莫(dipyridamole) 又名潘生丁(persantine),其抑制血小板功能主要是通过对血小板磷酸二脂酶抑制而升高cAMP,以致降低血小板粘连和聚集。并且它阻止红细胞及血管内皮细胞对腺苷(Ade)的摄取和代谢,以致增加了Ade在血浆中浓度,激活了腺苷环化酶,增加cAMP而抑制血小板聚集。双嘧达莫(DP)对脑卒中二级预防是否有效一直未加肯定。最近,肯定DP对脑卒中二级预防有效的一个临床试验是来自ESPS2的报告[21]。有13个欧洲国家参与试验,他们采用安慰剂对照和双盲试验,随机分成4个组:低剂量ASA 50 mg.d-1 (n=1649)组,DP缓释剂型组400 mg.d-1 (n=1654),ASA合并服用DP组(n=1650)以及安慰剂组(n=1649)。随诊2年,以脑卒中复发和死亡率为指标。与安慰剂组比较,发现卒中发病率分别降低18.1%(ASA),16.3%(DP)和37%(ASA+DP)。脑卒中发作和死亡率分别降低13.2%,15.4%和24.4%。从这一规模较大的试验结果可以看出DP是有效的,而且与ASA合并用药其疗效有增强作用。
, 百拇医药
噻氯匹定(ticlopidine) 是一新型的抗血小板药,它抑制血小板功能是通过抑制ADP及多种激动剂引起的血小板聚集。可能的机制是选择性干扰ADP诱发的纤维蛋白原与糖蛋白IIb/IIIa复合物的结合。对磷脂酶A活性及对AA代谢物均无影响。对cAMP-AC系统也无直接作用。一般口服后24~48 h开始起效,3~5 d作用最强,停药后药效尚能维持72 h。
有数个多中心、随机、双盲和安慰剂对照试验,噻氯匹定均能有效地预防TIA复发[24,25]。Bellavance报道一个多中心、双盲、随机临床试验表明,噻氯匹定剂量为250 mg.d-1 (n=1529),ASA为650 mg.d-1 (n=1540),随诊5.8年,噻氯匹定和ASA均明显降低脑卒中危象,但前者疗效略好。但由于价贵及较严重的中性粒细胞减少的副作用,因此主张适用于不能耐受ASA的病人,或在ASA治疗期间有缺血性脑卒中发作的患者。
, 百拇医药
根据上述抗血小板药的临床试验资料,凡患者有近期TIA史,或有轻度完全性脑卒中而CT未证实有脑出血病人均可用抗血小板药物进行二级预防。Matchar等[26]综述了自1977年来发表的应用抗栓药如ASA,DP,噻氯匹定或硫氧唑酮预防脑血管病的概况。选择随机和安慰剂对比临床试验材料为依据,得出以下结论:认为(1) TIA或轻度脑卒中患者ASA为首选药物;(2) 对ASA无反应或耐受者,而且在治疗期间发生脑卒中者可用噻氯匹定治疗;(3) 在接受抗凝治疗有出血危险因子者可选用ASA。
4 治疗脑血管病恢复期和后遗症的药物
这类药物的作用性质大多数为脑代谢和脑循环改善剂。有些药物有较好的药效学实验基础,而有的则较为粗糙,在我国临床上应用较多的药物有:
银杏叶提取物(Ginkgo biloba extracts,GBE)[27] 银杏是一种古老的植物,其果实为传统中药。自60年代始,西方国家从叶中提取并分离其成分。有药理活性的成份有两种:一类是黄酮类似物(flavonoids)另一类是银杏苦内酯(即ginkgolides,或terpenes),从中已分离出银杏苦苷A,B,C,J和M等单体。其中以银杏苦苷B(BN 52021)的作用最强,A(BN 52020)次之,余较弱。银杏提取物中主要含黄酮苷和银杏苦内酯。
, 百拇医药
其药理作用为:(1) 黄酮类似物有清除自由基,抑制脂质过氧化的作用。能扩张血管,增加脑血流量。(2) 银杏苦内酯则对PAF有特异性抑制作用,因此能对抗血小板聚集和防止血栓形成作用。(3) BN 52021能抑制脑组织内水、Na+蓄积和K+的丢失,减轻脑水肿的形成。(4) GBE对实验性局部脑缺血有一定保护作用,降低多发性脑梗塞大鼠BBB的损伤,使减轻脑水肿;能改善缺血脑ATP合成;对实验性引起兔SAH的血管痉挛呈明显的抑制作用。GBE的主要适应证为脑功能不全。
艾地苯醌(idebenone) 药理作用:(1) 改善脑功能障碍,(2) 改善脑能量代谢,(3) 对线粒体肿胀和脂质过氧化有抑制作用。可用于改善脑代谢和精神症状。
其他 丹参注射剂,丹参对实验性脑缺血研究较少,据报道丹参有抗体外血栓形成和抗血小板聚集功能,对收缩状态的微血管有显著的扩张作用,可改善微循环障碍,改善细胞缺血、缺氧所致的代谢障碍。临床试验认为有一定效果;长春西丁(vinpocetine,商品名为卡兰)为脑循环和脑代谢改善剂,对脑缺血有保护作用。用于治疗脑卒中后遗症,脑动脉硬化症。但尚缺乏严格的多中心,随机,双盲,安慰剂对照试验的检验。另有氢化麦角碱、川芎嗪、二苯美伦(befemelane)、环扁桃酯(cyclandelate,商品名为抗栓丸)、胞二磷胆碱(citilcholine)、已酮可可碱(pentoxifylline)和中药神农丸等的临床适应证较相似,多数适用于脑卒中恢复期和后遗症。
, 百拇医药
5 本实验室研究开发的新药
作者实验室从整体动物、细胞和分子生物学等不同水平,对抗脑缺血新药丁苯酞进行了较广泛而深入的研究。丁苯酞(dl-butylphthalide,NBP)是从芹菜籽中提取出来的左旋体,后经人工合成为消旋体。经研究发现NBP抗脑缺血的药效学作用如下:(1) NBP(150~200 mg.kg-1 sc)具有改善小鼠全脑缺血脑能量代谢,即能逆转缺血时ATP和PCr的降低和LA的升高,使其恢复和接近正常水平[28]。(2) NBP(10~20 mg.kg-1 ip)能增加局部脑缺血大鼠(MCAO)缺血区的局部脑血流(rCBF)[29]。(3) NBP(10~20 mg.kg-1 ip或40~160 mg.kg-1 po)能缩小MCAO大鼠脑梗塞面积和改善神经功能缺失[30]。(4) NBP(80~240 mg.kg-1 po)能减轻MCAO大鼠脑水肿[31],(5) NBP(30~100 mg.kg-1 po)能改善MCAO大鼠记忆功能障碍[32]。(6) NBP(100 mg.kg-1 po)对脑卒中易感型自发性高血压大鼠有预防和治疗作用[33]。(7) NBP(1~100 μmol.L-1)对多种原因如高钾、兴奋毒和低氧低糖引起的培养胎大鼠神经细胞损伤有直接保护作用[34]。
, http://www.100md.com
对NBP作用机制研究表明:(1) NBP(5~10 mg.kg-1 ip)对t-MCAO(暂时性MCAO)大鼠脑线粒体呼吸链复合酶IV活性抑制有明显逆转作用[35],提示改善缺血脑能量代谢(增加ATP,PCr,降低LA)是通过这一作用而发挥的;(2) NBP有降低大鼠尾动脉环细胞内钙的作用,而对外钙无影响[36],对神经细胞内钙也有明显降低作用(待发表资料);(3) 发现d-NBP可激活大鼠脑突触体胞液cNOS活性,增加NO和cGMP,对iNOS无作用,而l-NBP则能抑制cNOS和iNOS。随后的实验表明,蛛网膜下腔出血引起的rCBF降低,d-NBP能改善rCBF,而l-NBP则无影响。由此推测,NBP可能是通过激活cNOS而增加rCBF(待发表资料);(4) NBP(20~40 mg.kg-1 ip)能抑制全脑缺血(4血管阻断)大鼠纹状体细胞外液ATP降解代谢产物次黄嘌呤(Hyp)和黄嘌呤(Xan)的含量升高[37],提示O()/(.)2底物生成减少,降低了
的过量产生;(5) NBP(10~20 mg.kg-1 ip)能明显抑制t-MCAO大鼠脑皮层缺血再灌期AA含量的增加(待发表资料);(6) NBP能明显抑制局部脑缺血大鼠缺血再灌期脑MDA的产生,体外实验表明NBP能抑制黄嘌呤和黄嘌呤氧化酶所生成的(待发表资料);(7) NBP能降低有害刺激对胎大鼠培养神经细胞谷氨酸(Glu)释放的增加;(8) 能增加局部脑缺血大鼠缺血再灌期PGI2/TXA2的比值[38];(9) 用原位杂交法和Nothern印迹杂交法证明NBP能明显抑制局部脑缺血大鼠缺血再灌后12和24 h的hsp70mRNA的表达[39]。由上述结果可见,NBP作用于多个病理环节:如改善线粒体功能和脑微循环,降低Glu的释放,抑制[Ca2+]i和AA,抑制和MDA,特别是前两者是它发挥抗脑缺血的主要作用。由于局部脑缺血的脑损伤在始初和迟发性损伤过程中是一多因素参与的过程,如在治疗上只解决复杂过程中的一个方面,恐怕不会收到好的效果。因此在治疗上去阻断脑缺血病理过程的多个环节是很重要的。而NBP正是具有作用于多个病理环节的药物,这对其开发为抗脑缺血药物具有良好的前景。现在NBP正在进行II期临床试验。, http://www.100md.com
6 结语和展望
急性缺血性脑卒中严重危害着人类健康,幸存者往往留下后遗症,影响病人生活质量,也增加家庭和社会负担。为此,探索其病理生理,研究药物干预极为重要。自80年代以来,很多学者除提出能量代谢、兴奋毒、氧化损伤、钙超载等学说外、最近也注意到脑缺血与细胞因子、炎症反应、粘连分子等密切相关。相应的药物也应运而出,如钙拮抗剂、兴奋性受体拮抗剂和自由基清除剂等,但能否治疗急性缺血性脑卒中仍不能完全肯定。临床上用溶栓药如t-PA等治疗缺血性脑卒中(在发作后6 h内用药)虽收到较好效果,但尚未解决出血率高的危险性。由于缺乏治疗药物,现在各国多用预防药如阿司匹林等进行二级预防,取得较好效果。近年来,各国学者对缺血性脑损伤的病理生理进行了深入探讨,企图寻找一个能阻断缺血性脑损伤的多个病理环节的新药,相信这一天将会到来。
关键词 脑缺血;一氧化氮;细胞因子;炎症反应;丁基苯酞
联系人 Tel:(010)63165173, Fax:(010)63017757,E-mail:fengyp@public.gb.com.cn
, 百拇医药
参考文献
1 李世绰.中国循环杂志,1993,8∶577
2 冯亦璞.陈维洲等主编.心血管药理学进展.北京:人民卫生出版社,1995.90
3 Siesjo BK, Katsura KI, Zhao Q, et al. J Neurotrauma, 1995,12∶943
4 MacManus JP, Linnik MD. J Cereb Blood Flow Metab, 1997,17∶815
5 Beckman JS, Ye YZ, Chen J, et al. Advances in Neurology. Vol 71. In: Siesjo Bk, Wieloch T, ed. Philadelphia: Lippinocott-Raven Publihers, 1996.339
, 百拇医药
6 The RANTTAS Investigators. Stroke, 1996,27∶1453
7 Faraci FM, Brain JE. Stroke, 1994,25∶692
8 Siesjo Bk, Wieloch T, ed. Advances in Neurology. Vol 71. Philadelphia: Lippinocott-Raven Pubblihers, 1996.355
9 Samdani AF, Dawson TM, Dawson VL. Stroke, 1997,28∶1283
10 Iadecola C. Trends Neurosci, 1997,20∶132
11 Tomita M, Fukuuchi Y. Acta Neurochim, 1996,66(Suppl)∶32
, 百拇医药
12 Kim JS. J Neurosci, 1996,137∶67
13 The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. N Engl J Med, 1995,333∶1581
14 Hacke W, Kaste M, Fieschi C, et al. J Am Med Assoc, 1995,274∶1017
15 Pickard JD, Murray GD, Illingworth R, et al. Br Med J, 1989,298∶636
16 Harders A, Kakarieka A, Braakman R, et al. J Neurosurg, 1996,85∶82
, http://www.100md.com
17 The American Nimodipine Study Group. Stroke, 1992,23∶3
18 Kaste M, Fogelholm R, Erila T. Stroke, 1994,25∶1348
19 汪钟,郑植荃主编.现代血栓病学.北京:北京医科大学中国协和医科大学联合出版社,1997.508
20 The SALT Collaborative Group. Lancet, 1991,338∶1345
21 Diener HC, Cunha L, Forbes C, et al. J Neurol Sci, 1996,143∶1
22 CAST (Chinese Acute Stroke Trial) Collaborative group. Lancet, 1997,349∶1641
, http://www.100md.com
23 International Stroke Trial Collaborative Group. Lancet, 1997,349∶1569
24 Gent M, Blakely JA, Easton JD, et al. Lancet, 1989,1∶1215
25 Bellavance A. Stroke, 1993,24∶1452
26 Matchar DB, McCrory DC, Barnett HJ, et al. Ann Inter Med, 1994,121∶41
27 陈维洲,芮耀诚主编.脑血管病基础与临床研究.济南:山东科学技术出版社,1993∶153
28 冯亦璞,胡盾,张丽英.药学学报,1995,30∶741
, 百拇医药
29 Yan CH, Feng YP, Zhang JT. Acta Pharmacol Sin, 1998,19∶117
30 刘小光,冯亦璞.药学学报,1995,30∶896
31 Deng WB, Feng YP. Chin Med Sci J, 1997,12∶102
32 胡盾,张丽英,冯亦璞.中国药理学与毒理学杂志,1997,11∶14
33 张丽英,冯亦璞.药学学报,1996,31∶18
34 阎超华,张均田,冯亦璞.药学学报,1997,32∶340
35 熊杰,冯亦璞.药学学报,1999,34∶(印刷中)
, 百拇医药
36 刘小光,冯亦璞.中国药理学与毒理学杂志,1996,10∶113
37 胡盾,黄新祥,冯亦璞.药学学报,1996,31∶13
38 Chong ZZ, Feng YP. Acta Pharmacol Sin, 1997,18∶505
39 熊杰,冯亦璞.药学学报,1998,33∶401
PATHOPHYSIOLOGY OF ISCHEMIC STROKE
AND STATUS OF DRUG INTERVENTION
Feng Yipu(Feng YP)
(Institute of Materia Medica, Chinese Academy of Medical Sciences
and Peking Union Medical College, Beijing 100050, China)
KEY WORDS cerebral ischemia; nitric oxide; cytokine; inflammatory reaction; dl-butyphthalide
收稿日期: 1998-11-10, 百拇医药