两种PCR技术在GDNF基因克隆中的应用
作者:崔振中 张淑琴 王晓民 韩济生
单位:北京医科大学神经科学研究所,北京 100083
关键词:
北京医科大学学报990129 聚合酶链式反应(polymerase chain reaction, PCR)自1985年问世以来在分子生物学领域得到了极其广泛的应用,包括从基因组中大量地扩增单拷贝基因[1,2],扩增mRNA的RT-PCR[3],PCR测序,PCR标记探针,定量PCR[4],反向PCR及临床基因诊断等许多方面。本文报道在GDNF基因克隆过程中2种新的PCR技术在直接基因克隆和重组体方向鉴定方面的应用。
Ⅰ 一步克隆法构建GDNF基因表达载体
用PCR方法构建了pcDNA3-T表达载体,将通过RT-PCR扩增获得的人GDNF基因直接克隆到表达载体中,使该基因可以直接在真核细胞中进行表达。
, http://www.100md.com
1 材料与方法
GDNF基因(cDNA),本室克隆[5];DH5α菌种及pcDNA3质粒,本室保存;dNTPs,Taq DNA聚合酶,T4 DNA连接酶,EcoRⅤ,购自Promege公司。
pcDNA3质粒的酶切:pcDNA3质粒5 μg,EcoRⅤ(10u.μl-1)1 μl,Buffer D2 μl,加H2O至20 μl,37℃保温2 h。酶切结束后取1 μl酶切产物进行琼脂糖凝胶电泳检测酶切结果,酶切完全后补加水至100 μl,酚及酚/氯仿抽提,乙醇沉淀,重溶于10 μl水中。
构建T-克隆载体:取一个1.5 ml的小离心管,依次加入EcoRⅤ酶切的平端pcDNA3质粒10 μl,10×Taq DNA聚合酶buffer 10 μl,50 mmol.L-1 MgCl2 10 μl,10 mmol.L-1 dTTP 10 μl,Taq DNA聚合酶(5 u.μl-1)1 μl,加H2O至100 μl,覆盖50 μl石蜡油,于PCR循环仪上75 ℃保温2 h,扩增结束后用酚及酚/氯仿抽提,乙醇沉淀,重溶于100 μl水中,-20℃保存。
, 百拇医药
连接反应:取T-克隆载体1 μl(50 ng),加入2 μl(100 ng)GDNF基因PCR产物,T4连接酶Buffer 1 μl,T4 DNA连接酶(10 u.μl-1)0.5 μl,加水至10 μl,16 ℃连接过夜。取连接产物5 μl转化大肠杆菌DH5α感受态菌,筛选重组克隆。
2 结果
经克隆筛选及HindⅢ/BamHⅠ双酶切分析,获得了多个重组克隆,重组质粒的酶切分析结果见图1。表明利用本方法构建的pcDNA3-T载体系统可顺利地克隆PCR产物。重组质粒的方向鉴定方法见本文Ⅱ。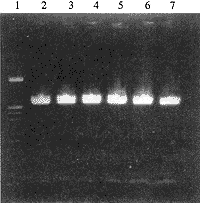
1,λ DNA HindⅢ/EcoRⅠ marker (from top to bottom are 21 227,5 148 and 4 973,4 269,3 530,2 027,1 904,1 587,1 375,947,831,564 bp); 2-7,represent different clones.
, 百拇医药
图1 重组质粒的酶切分析
Figure 1 Restriction analysis of recombinant plasmids
3 讨论
由于Taq DNA聚合酶具有在PCR产物的3′端非特异地加一个A的特性[6],使PCR产物的直接平端克隆产生一定困难。传统的做法是用商品化的pGEM-T系列载体,将PCR产物克隆,如需要则进行序列分析,再用载体上提供的酶切位点将目的基因重新克隆于特定的表达载体上。因此在操作上需要重复克隆,费时费力,而且商品化的T-载体所提供的酶切位点终究有限,在构建表达载体时方向的选择往往不合适。采用本文的方法,在选定的真核或原核表达载体上用能产生平端的内切酶如HincⅡ、EcoRV、SmaⅠ、PvuⅡ等进行消化,使产生的平端正好位于载体提供的启动子下游,用PCR方法只提供dTTP,由Taq DNA聚合酶在3′端补加一个T的接头,与经PCR扩增获得的目的基因连接,经方向鉴定后可直接用于表达。用此方法可为实验者提供多种选择,避免了商品化T-载体的缺陷,同时还可以节省经费,达到事半功倍的效果。
, 百拇医药
Ⅱ 用PCR方法鉴定重组克隆的方向
设计了一对PCR引物,可以直接对上述克隆进行方向鉴定,从而获得插入方向正确的重组克隆直接用于真核细胞的表达。
1 材料与方法
PCR引物:上游引物:5′TGG AAT TCT GCA GAT TTA TG;下游引物:5′CGC CAC TGT GCT GGA TTT CA 由Sybesyn公司合成。其它材料同本文Ⅰ。重组克隆方向鉴定:用上述重组克隆(图1)为模板进行PCR反应。PCR反应条件:94 ℃ 2 min;94 ℃ 50 s,56 ℃ 50 s,72 ℃ 90 s,30个循环;72 ℃ 5 min。琼脂糖电泳检测PCR扩增结果。
2 结果
经PCR检测在上述克隆中获得了2个插入方向正确的重组克隆,从PCR扩增的结果看有预期的558 bp片段的扩增。PCR检测结果见图2。选取其中一个克隆定名为pCMV/hGDNF,经系列酶切分析证实这是一个正确的重组克隆,其酶切分析图谱未给出。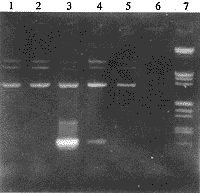
, 百拇医药
1,2,5,negative recombinant clones (with reverse direction of GDNF gene); 3,4,two positive recombinant clones (with forward direction of GDNF gene); 6,negative control of PCR; 7,λ DNA HindⅢ/EcoRⅠ marker (from top to bottom are 21 227,5 148 and 4 973,4 269,3 530, 2 027, 1 904,1 587,1 375,947,831,564 bp).
图2 用PCR方法鉴定重组克隆的方向
Figure 2 Determination of the direction of recombinant plasmids by PCR
3 讨论
, 百拇医药
在构建目的基因的表达载体时最困难的步骤是重组克隆的筛选,而表达载体的构建往往没有蓝/白筛选标记,用平端或单个碱基的粘端克隆时,目的基因的插入方向是不确定的,用酶切分析可以大致确定重组体的方向,但有时因为酶切位点的限制很难确定重组体的方向。我们设计的PCR上游引物有16个碱基与载体匹配,4个碱基与目的基因的5′端匹配;下游引物同样有16个碱基与载体匹配,4个碱基与目的基因的3′端匹配。如果重组体是按正确的方向插入到载体中,则有PCR产物扩增,如果重组体是反向插入则没有PCR产物扩增。这样可以非常方便地鉴定重组体的方向,此法可靠、快捷,并可避免因某些酶切分析所造成的假象。
本文报道了两种PCR技术在GDNF基因克隆过程中的思考与运用,或许会给相关人员一点启发。
参考文献
1 Saiki RK, Scharf S, Faloona F, et al. Enzymatic amplification of globulin sequences and restriction site analysis for diagnosis of sickle cell anemia. Science, 1985,230:1350-1354
, http://www.100md.com
2 Saiki RK, Gelfand DH, Stoflel S, et al. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science, 1989,239:487-490
3 Berchtode MW. A simple method for direct cloning and sequencing cDNA by the use of a single specific oligo (dT) in a polymeraes chain reaction (PCR). Nucl Acids Res, 1989,17:453-457
4 Katz JP,bodin ET, Coen DM. Quantitative polymerase chain reaction analysis of herpes simplex virus DNA ganglia of mice infected with replication-incompetent mutants. J Virol, 1990,64:4288-4295
5 崔 巍,崔振中,王晓民,等.人GDNF基因的克隆,北京医科大学学报, 1998,30:15-17
6 Clark JM. Novel nontemplated nucleotide addition reactions catalyzed by prokaryotic and eukaryotic DNA polymerase. Nucl Acids Res, 1988,16:9677-9686
(1998-03-30收稿), http://www.100md.com
单位:北京医科大学神经科学研究所,北京 100083
关键词:
北京医科大学学报990129 聚合酶链式反应(polymerase chain reaction, PCR)自1985年问世以来在分子生物学领域得到了极其广泛的应用,包括从基因组中大量地扩增单拷贝基因[1,2],扩增mRNA的RT-PCR[3],PCR测序,PCR标记探针,定量PCR[4],反向PCR及临床基因诊断等许多方面。本文报道在GDNF基因克隆过程中2种新的PCR技术在直接基因克隆和重组体方向鉴定方面的应用。
Ⅰ 一步克隆法构建GDNF基因表达载体
用PCR方法构建了pcDNA3-T表达载体,将通过RT-PCR扩增获得的人GDNF基因直接克隆到表达载体中,使该基因可以直接在真核细胞中进行表达。
, http://www.100md.com
1 材料与方法
GDNF基因(cDNA),本室克隆[5];DH5α菌种及pcDNA3质粒,本室保存;dNTPs,Taq DNA聚合酶,T4 DNA连接酶,EcoRⅤ,购自Promege公司。
pcDNA3质粒的酶切:pcDNA3质粒5 μg,EcoRⅤ(10u.μl-1)1 μl,Buffer D2 μl,加H2O至20 μl,37℃保温2 h。酶切结束后取1 μl酶切产物进行琼脂糖凝胶电泳检测酶切结果,酶切完全后补加水至100 μl,酚及酚/氯仿抽提,乙醇沉淀,重溶于10 μl水中。
构建T-克隆载体:取一个1.5 ml的小离心管,依次加入EcoRⅤ酶切的平端pcDNA3质粒10 μl,10×Taq DNA聚合酶buffer 10 μl,50 mmol.L-1 MgCl2 10 μl,10 mmol.L-1 dTTP 10 μl,Taq DNA聚合酶(5 u.μl-1)1 μl,加H2O至100 μl,覆盖50 μl石蜡油,于PCR循环仪上75 ℃保温2 h,扩增结束后用酚及酚/氯仿抽提,乙醇沉淀,重溶于100 μl水中,-20℃保存。
, 百拇医药
连接反应:取T-克隆载体1 μl(50 ng),加入2 μl(100 ng)GDNF基因PCR产物,T4连接酶Buffer 1 μl,T4 DNA连接酶(10 u.μl-1)0.5 μl,加水至10 μl,16 ℃连接过夜。取连接产物5 μl转化大肠杆菌DH5α感受态菌,筛选重组克隆。
2 结果
经克隆筛选及HindⅢ/BamHⅠ双酶切分析,获得了多个重组克隆,重组质粒的酶切分析结果见图1。表明利用本方法构建的pcDNA3-T载体系统可顺利地克隆PCR产物。重组质粒的方向鉴定方法见本文Ⅱ。
1,λ DNA HindⅢ/EcoRⅠ marker (from top to bottom are 21 227,5 148 and 4 973,4 269,3 530,2 027,1 904,1 587,1 375,947,831,564 bp); 2-7,represent different clones.
, 百拇医药
图1 重组质粒的酶切分析
Figure 1 Restriction analysis of recombinant plasmids
3 讨论
由于Taq DNA聚合酶具有在PCR产物的3′端非特异地加一个A的特性[6],使PCR产物的直接平端克隆产生一定困难。传统的做法是用商品化的pGEM-T系列载体,将PCR产物克隆,如需要则进行序列分析,再用载体上提供的酶切位点将目的基因重新克隆于特定的表达载体上。因此在操作上需要重复克隆,费时费力,而且商品化的T-载体所提供的酶切位点终究有限,在构建表达载体时方向的选择往往不合适。采用本文的方法,在选定的真核或原核表达载体上用能产生平端的内切酶如HincⅡ、EcoRV、SmaⅠ、PvuⅡ等进行消化,使产生的平端正好位于载体提供的启动子下游,用PCR方法只提供dTTP,由Taq DNA聚合酶在3′端补加一个T的接头,与经PCR扩增获得的目的基因连接,经方向鉴定后可直接用于表达。用此方法可为实验者提供多种选择,避免了商品化T-载体的缺陷,同时还可以节省经费,达到事半功倍的效果。
, 百拇医药
Ⅱ 用PCR方法鉴定重组克隆的方向
设计了一对PCR引物,可以直接对上述克隆进行方向鉴定,从而获得插入方向正确的重组克隆直接用于真核细胞的表达。
1 材料与方法
PCR引物:上游引物:5′TGG AAT TCT GCA GAT TTA TG;下游引物:5′CGC CAC TGT GCT GGA TTT CA 由Sybesyn公司合成。其它材料同本文Ⅰ。重组克隆方向鉴定:用上述重组克隆(图1)为模板进行PCR反应。PCR反应条件:94 ℃ 2 min;94 ℃ 50 s,56 ℃ 50 s,72 ℃ 90 s,30个循环;72 ℃ 5 min。琼脂糖电泳检测PCR扩增结果。
2 结果
经PCR检测在上述克隆中获得了2个插入方向正确的重组克隆,从PCR扩增的结果看有预期的558 bp片段的扩增。PCR检测结果见图2。选取其中一个克隆定名为pCMV/hGDNF,经系列酶切分析证实这是一个正确的重组克隆,其酶切分析图谱未给出。
, 百拇医药
1,2,5,negative recombinant clones (with reverse direction of GDNF gene); 3,4,two positive recombinant clones (with forward direction of GDNF gene); 6,negative control of PCR; 7,λ DNA HindⅢ/EcoRⅠ marker (from top to bottom are 21 227,5 148 and 4 973,4 269,3 530, 2 027, 1 904,1 587,1 375,947,831,564 bp).
图2 用PCR方法鉴定重组克隆的方向
Figure 2 Determination of the direction of recombinant plasmids by PCR
3 讨论
, 百拇医药
在构建目的基因的表达载体时最困难的步骤是重组克隆的筛选,而表达载体的构建往往没有蓝/白筛选标记,用平端或单个碱基的粘端克隆时,目的基因的插入方向是不确定的,用酶切分析可以大致确定重组体的方向,但有时因为酶切位点的限制很难确定重组体的方向。我们设计的PCR上游引物有16个碱基与载体匹配,4个碱基与目的基因的5′端匹配;下游引物同样有16个碱基与载体匹配,4个碱基与目的基因的3′端匹配。如果重组体是按正确的方向插入到载体中,则有PCR产物扩增,如果重组体是反向插入则没有PCR产物扩增。这样可以非常方便地鉴定重组体的方向,此法可靠、快捷,并可避免因某些酶切分析所造成的假象。
本文报道了两种PCR技术在GDNF基因克隆过程中的思考与运用,或许会给相关人员一点启发。
参考文献
1 Saiki RK, Scharf S, Faloona F, et al. Enzymatic amplification of globulin sequences and restriction site analysis for diagnosis of sickle cell anemia. Science, 1985,230:1350-1354
, http://www.100md.com
2 Saiki RK, Gelfand DH, Stoflel S, et al. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science, 1989,239:487-490
3 Berchtode MW. A simple method for direct cloning and sequencing cDNA by the use of a single specific oligo (dT) in a polymeraes chain reaction (PCR). Nucl Acids Res, 1989,17:453-457
4 Katz JP,bodin ET, Coen DM. Quantitative polymerase chain reaction analysis of herpes simplex virus DNA ganglia of mice infected with replication-incompetent mutants. J Virol, 1990,64:4288-4295
5 崔 巍,崔振中,王晓民,等.人GDNF基因的克隆,北京医科大学学报, 1998,30:15-17
6 Clark JM. Novel nontemplated nucleotide addition reactions catalyzed by prokaryotic and eukaryotic DNA polymerase. Nucl Acids Res, 1988,16:9677-9686
(1998-03-30收稿), http://www.100md.com