Duchenne/Becker肌营养不良症基因缺失的检测及缺失类型与临床表型的关系
作者:刘玉阁 刘辉 谢丙
单位:300052 天津医科大学总医院神经病学研究所
关键词:聚合酶链反应;肌营养不良;基因缺失
中华儿科杂志/980412 【摘要】 目的 了解Duchenne/Becker肌营养不良症(DMD/BMD)基因缺失的分布及其与表型的关系。 方法 采用18对引物多重聚合酶链反应(mPCR)分两步对96例DMD/BMD进行筛查。 结果 用第一组引物检出49例缺失,第二组引物检出11例缺失,分别占全长cDNA探针检出缺失的79%和18%,两组引物检出缺失的总和占全部缺失的97%。60例缺失中,DMD42例(70%),中间型3例(5%),BMD13例(22%),未分类2例(3%)。43例(72%)缺失分布于外显子44~52,17例(28%)分布于外显子1~19。结论 基因缺失主要分布于基因3′侧外显子44~52和5′侧外显子12~19两个热区。临床表型与基因缺失类型有密切关系,41例移码缺失破坏了翻译阅读框架而导致基因功能丧失,表型为严重的DMD,11例整码缺失位于外显子2~43,基因部分功能保留,表型为较轻的中间型和BMD。
, http://www.100md.com
Detection of Duchenne/Becker muscular dystrophy (DMD/BMD) gene deletions and its association with phenotype
Liu Yuge, Liu Hui, Xie Bingdi.
Neurological Research Institute, General Hospital, Tianjin Medical University, Tianjin 300052
【Abstract 】 Objective To analyse the deletion distribution and the relation of distribution of gene deletions and phenotype in Duchenne/Becker muscular dystrophy (DMD/BMD). Methods Ninety-six patients with DMD/BMD were screened with the multiplex polymerase chain reaction (mPCR) amplification using 18 pairs of primers. Results Forty-nine deletions were detected with the first set and 11 with the second set which accounted for 79% and 18% of those detected by full length cDNA probes respectively. These two multiplex reactions detected about 97% of deletions in DMD/BMD patients. In 60 deletions, 42 were of DMD (70%), 3 intermediate (5%), 13 BMD (22%), and 2 cases (3%) were not classified. Forty-three deletions (72%) were located in exons 44~52 and 17 (28%) in exons 1~19. Conclusions The gene deletions were mainly distributed in the 3′ terminus of the gene central region around exons 44~52 and 5′ terminus of the gene around exons 12~19. The phenotype was associated with the type of gene deletion in DMD/BMD. The phenotype of 41 frameshift deletions was severe DMD, which disrupted the reading frame, resulting in the loss of the gene function. The phenotype of 11 in -frame deletions distributed in exons 2~43 with partial gene functions was intermediate patient and BMD.
, 百拇医药
【Key words 】 Polymerase chain reaction Muscular dystrophy Gene deletion
Duchenne/Becker肌营养不良症(DMD/BMD)是一种常见的X连锁隐性遗传性疾病,二者是等位基因疾病,发病率分别为新生男婴的1/3 500和1/30 000。1/3由新生突变引起[1]。近年对DMD/BMD的研究取得了新的进展,致病基因已定位于Xp21,基因长2 300kb,含70个外显子,编码一种位于肌细胞膜上的骨架蛋白-“肌营养不良蛋白”(dystrophin),基因缺失或重复均导致进行性肌营养不良症[2,3]。Chamberlain等[4]采用9对引物多重聚合酶链反应(mPCR)检出DMD/BMD基因缺失的80%,Beggs等[5]设计了另外9对引物并联合应用这两组引物检出基因缺失的98%,从而对本病基因检测建立了一种简便快速的诊断方法。本组参照上述引物序列合成18对引物,对我院经临床,肌电图,血清酶学和肌活检确诊的96例无亲缘关系的DMD/BMD进行基因缺失筛查,并对我国DMD/BMD基因缺失的分布及其与临床表型的关系进行初步探讨。
, 百拇医药
对象和方法
一、对象
96例患者均为我院1992~1996年住院或门诊患者,均为男性,发病年龄6~18岁(平均12.4±3.3岁),根据丧失行走能力的年龄将患者分为4型:(1)Duchenne型(DMD)65例(68%),8~13岁以前丧失行走能力;(2)中间型6例(6%),13~16岁丧失行走能力;(3)Becker型(BMD)20例(21%),16岁后仍能行走;(4)5例(5%)8岁以下患者未能分型。临床均表现为四肢近端对称性进行性加重的肌肉萎缩,70%伴有腓肠肌或二头肌假肥大,均有Gower征阳性。肌电图检查显示典型肌源性改变,血清肌酸磷酸激酶(CK)及其同功酶MCK均显著增高,肌活检显示镶嵌分布的肌纤维萎缩,肥大,玻璃样变和坏死等典型病理改变。30例正常对照均为非神经肌病患者。
二、测定方法
1.DNA的提取:抽取患者和正常对照外周血5 ml,加入0.2%依地酸,乙二胺四乙酸(EDTA)抗凝,采用等渗溶血法裂解红细胞后离心分离白细胞,加入1%十二烷基硫酸钠(SDS)和蛋白酶K,37℃消化4~6小时,再加入1/3体积的饱和氯化钠,离心除去沉淀,用等体积的氯仿抽提两次,2.5倍体积的无水乙醇沉淀,75%乙醇漂洗,晾干后溶于TE(10 mmol/L Tris.CI, 1 mmol/L EDTA,pH 7.6),-20℃保存。
, 百拇医药
2.mPCR:18对引物参照Chamberlain和Beggs等[4,5]提供的序列由美国CyberSyn公司合成,经寡核苷酸纯化柱(OPC)纯化。将18对引物分为两组(第1组:外显子4,8,12,17,19,44,45,48,51;第2组:启动子,外显子3,6,13,43,47,50,52,60),分两步对全组患者进行mPCR扩增。50 μl PCR反应体系含670 mmol Tris-HCl(pH 8.0),166mmol(NH4)2SO4,67 mmol MgCl2,10 mmol β-巯基乙醇,10 μg/μl小牛血清白蛋白(BSA),25 mmol dNTP,每对引物各0.25 μmol,模板DNA 500 ng。将反应液97℃予变性10分钟,加入5UTaqDNA聚合酶(德国BM产品),混匀后加入35 μl石蜡油。按照94℃变性30秒,56℃复性30秒,70℃延伸2分钟进行30次循环,最后70℃延伸7分钟。取10 μl扩增产物用2%琼脂糖或6%聚丙烯酰胺凝胶在0.5×TBE中电泳,溴化已锭染色,在紫外灯下观察结果并摄影,或用0.2%硝酸银染色保留结果。
, http://www.100md.com
结果
96例患者用18对引物mPCR扩增共检出60例缺失,其中用第1组引物检出49例缺失,检出率为51%,用第2组引物在未检出缺失的47例中检出11例缺失,检出率为12%(图1,2)。两组引物检出的总缺失率为63%。60例缺失中DMD42例(70%),中间型3例(5%),BMD13例(22%),未分类2例(3%);41例为移码缺失,19例为整码缺失。全部缺失均经全长cDNA探针(1-2,2b-3,4-5a,5b-7,8,9-14)(质粒由Kunkel教授赠送)Southern杂交证实,用18对引物检出的缺失占全长cDNA探针检出缺失的97%。因两组引物均未覆盖基因中心区,用cDNA探针4-5a和5b-7在这一区域内检出的两例缺失漏检,漏检率为3%。本组用外显子60未检出缺失,可能与病例数较少有关。两步mPCR检出基因3′侧外显子44~52缺失43例,占全部缺失的72%,5′侧外显子1~19缺失17例,占28%。30例正常对照均未见基因缺失。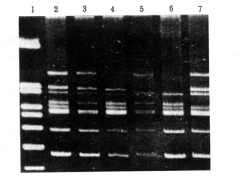
, http://www.100md.com
1:扩增带大小标志(PBR322HinfI);2:正常对照;3~7:DMD/BMD基因内缺失图1 第1组引物mPCR扩增DNA分析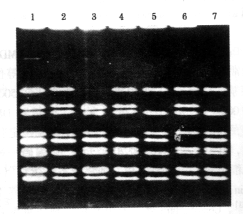
1:正常对照;2~7:DMD/BMD不同基因内缺失
图2 第2组引物mPCR扩增DNA分析
讨论
一、应用mPCR技术
mPCR技术的应用对于DNA分析是一种革命性的探索。以往DMD/BMD基因缺失需要使用七个不同的亚克隆cDNA探针检测,基因组DNA至少需用两种不同的限制性内切酶消化,检测步骤极其繁琐。应用mPCR技术大大检化了Southern印迹分析,根据大部分缺失集中在两个区域的理论,仅检测70个外显子中的一部分即可检测出大部分缺失。Chamberlain等[4]首先在基因3′侧和5′侧以9个缺失热点外显子(4,8,12,17,19,44,45,48,51)的旁侧序列设计了9对引物,一次mPCR扩增即可检出用全长cDNA探针检出缺失的80%。
, 百拇医药
二、应用引物扩增
Beggs等[5]在mPCR研究的基础上选择另外8个缺失热点外显子(3,6,13,43,47,50,52,60)和启动子设计了相应的9对引物,进一步扩大了缺失的检测范围,使本病基因缺失检出率达到98%以上。上述两组引物联合应用不仅能扩增外显子44~52缺失热区内的大部分外显子,而且还能检测5′端和基因远端的缺失,并可分析突变对翻译阅读框架的影响。
三、与Southern印迹分析比较
本组应用18对引物对96例DMD/BMD进行两步mPCR扩增结果与Southern印迹分析的结果一致。第1组引物检出49例缺失,占全长cDNA探针检出缺失的79%,第2组引物检出11例缺失,占全长cDNA探针检出缺失的18%,两组引物检出缺失的总和占Southern印迹分析检出缺失的97%。这一结果与国外文献报道的缺失检出率接近[5,6]。实践证明这两组引物也适用于中国人DMD的基因诊断。应该指出的是,应用mPCR可以避免Southern杂交在基因诊断中的某些失误,例如两例应用cDNA8探针杂交显示缺失外显子48~52,但是用mPCR扩增时外显子51和52扩增带均显示清晰。Southern杂交出现失误的原因可能是由于DNA印迹转移不良,或在膜杂交的过程中出现气泡阻碍杂交过程。由于两套引物均未覆盖基因中心区,本组两例相当cDNA探针4-5a和5b-7检测区域内外显子20~40和21~42缺失漏检,漏检率仅为3%。但由于mPCR快速,准确,基因组DNA用量少,采用两套引物即可覆盖基因3′和5′侧大部分缺失热区外显子和基因的远侧端,其优越性已大大超过Southern印迹分析。mPCR对反应条件要求较高,使用国产Taq酶有时出现假阳性。改用BM(德国)Taq酶,在缓冲液中加入0.01%明胶,并将循环次数增加至30~35次,可稳获9条扩增带。还应避免基因组DNA保存时间过长,以免DNA发生降解而出现假阳性。
, 百拇医药
四、DMD/BMD基因缺失分布规律
本组资料分析显示,DMD/BMD基因缺失的分布确有一定的规律。60例缺失中43例(72%)分布于外显子44~52,相当于cDNA探针8和7的3′侧4个外显子覆盖的区域内。基因缺失范围的大小不尽相同,其中以外显子48,50,51和52受累最多见。17例(28%)缺失分布于5′侧外显子1~19,其中以外显子12~19受累最多见,1~9次之。这种缺失分布不均一的现象表明DMD/BMD的缺失热区主要分布在基因中心区3′侧外显子44~52或48~52,另一个缺失热区分布在基因5′侧外显子12~19。Koenig等[7]通过对329例DMD/BMD Southern印迹分析,发现其中75%的基因缺失分布在基因中心区3′侧,25%分布在基因5′侧,并将这两个区域称为缺失热区。这一基因缺失分布规律相继被以后的研究所证实[5,6,8]。由此可见,我国DMD/BMD基因缺失的分布规律与欧洲白种人基本相似。
, 百拇医药
五、DMD/BMD的临床表型与基因缺失类型的关系
近年研究证实,DMD/BMD的临床表型与基因缺失类型有密切的关系[7]。本组41例患者为移码缺失,因缺失破坏了翻译阅读框架而导致基因功能丧失,临床表型为严重的DMD,常于9~13岁丧失行走能力。19例为整码缺失,其中11例缺失位于外显子2~43,因缺失部位不是基因的关键部位,部分基因功能得以保留,临床表型为症状较轻的中间型和BMD,分别于13~16岁和16岁以后丧失行走能力;8例缺失位于基因中心区3′侧,缺失均累及外显子51,其中6例(75%)表现为严重的DMD,仅两例(25%)为轻型BMD。Koenig等[9]发现51号外显子恰与肌细胞膜骨架蛋白dystrophin绞链3相对应,绞链3对维持dystrophin的弹性和柔韧性起着重要作用。故累及基因中心区3′侧外显子51的整码缺失临床多为严重的DMD表型。
参考文献
, 百拇医药
1 Koenig M, Hoffman EP, Bertelson CJ, et al. Complete cloning of the Duchenne muscular dystrophy cDNA. Cell, 1987, 50:509-517.
2 Monaco AP, Bertelson CJ, Liechti-Callati S, et al. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 1988, 2:90-95.
3 Burmeister M, Monaco AP, Gillard EF, et al. A 10-megabase physical map of human Xp21, including the Duchenne muscular dystrophy gene. Genomics, 1988, 2:189-202.
, http://www.100md.com
4 Chamberlain JS, Gibbs RA, Ranier HE, et al. Multiplex PCR for the diagnosis of Duchenne muscular dystrophy. New York: Acade Press, 1990. 272-281.
5 Beggs AH, Koenig M, Frederick M, et al. Detection of 98% of DMD/BMD gene deletions by polymerase chain reaction. Hum Genet, 1990, 86:45-48.
6 Abbs S, Yau SC, Clark S, et al. A convenient multiplex PCR system for the detection of dystrophin gene deletions. J Med Genet, 1991, 28:304-311.
, 百拇医药
7 Koenig M, Beggs AH, Moyer M, et al. The molecular basis for Duchenne versus Becker muscular dystrophy: correlation of severity with type of deletion. Am J Hum Gennet, 1989, 45:489-506.
8 Patino A, Narbona J, Garcia-Delgado M. Molecular analysis of the Duchenne muscular dystrophy gene in Spanish individuals. Am J Med Genet, 1995, 59:182-187.
9 Koenig M, Kunkel LM. Detailed analysis of the repeat domain of dystrophin reveals four potential hinge segments that may confer flecibility. J Bio Chem, 1990, 256:4560-4566., 百拇医药
单位:300052 天津医科大学总医院神经病学研究所
关键词:聚合酶链反应;肌营养不良;基因缺失
中华儿科杂志/980412 【摘要】 目的 了解Duchenne/Becker肌营养不良症(DMD/BMD)基因缺失的分布及其与表型的关系。 方法 采用18对引物多重聚合酶链反应(mPCR)分两步对96例DMD/BMD进行筛查。 结果 用第一组引物检出49例缺失,第二组引物检出11例缺失,分别占全长cDNA探针检出缺失的79%和18%,两组引物检出缺失的总和占全部缺失的97%。60例缺失中,DMD42例(70%),中间型3例(5%),BMD13例(22%),未分类2例(3%)。43例(72%)缺失分布于外显子44~52,17例(28%)分布于外显子1~19。结论 基因缺失主要分布于基因3′侧外显子44~52和5′侧外显子12~19两个热区。临床表型与基因缺失类型有密切关系,41例移码缺失破坏了翻译阅读框架而导致基因功能丧失,表型为严重的DMD,11例整码缺失位于外显子2~43,基因部分功能保留,表型为较轻的中间型和BMD。
, http://www.100md.com
Detection of Duchenne/Becker muscular dystrophy (DMD/BMD) gene deletions and its association with phenotype
Liu Yuge, Liu Hui, Xie Bingdi.
Neurological Research Institute, General Hospital, Tianjin Medical University, Tianjin 300052
【Abstract 】 Objective To analyse the deletion distribution and the relation of distribution of gene deletions and phenotype in Duchenne/Becker muscular dystrophy (DMD/BMD). Methods Ninety-six patients with DMD/BMD were screened with the multiplex polymerase chain reaction (mPCR) amplification using 18 pairs of primers. Results Forty-nine deletions were detected with the first set and 11 with the second set which accounted for 79% and 18% of those detected by full length cDNA probes respectively. These two multiplex reactions detected about 97% of deletions in DMD/BMD patients. In 60 deletions, 42 were of DMD (70%), 3 intermediate (5%), 13 BMD (22%), and 2 cases (3%) were not classified. Forty-three deletions (72%) were located in exons 44~52 and 17 (28%) in exons 1~19. Conclusions The gene deletions were mainly distributed in the 3′ terminus of the gene central region around exons 44~52 and 5′ terminus of the gene around exons 12~19. The phenotype was associated with the type of gene deletion in DMD/BMD. The phenotype of 41 frameshift deletions was severe DMD, which disrupted the reading frame, resulting in the loss of the gene function. The phenotype of 11 in -frame deletions distributed in exons 2~43 with partial gene functions was intermediate patient and BMD.
, 百拇医药
【Key words 】 Polymerase chain reaction Muscular dystrophy Gene deletion
Duchenne/Becker肌营养不良症(DMD/BMD)是一种常见的X连锁隐性遗传性疾病,二者是等位基因疾病,发病率分别为新生男婴的1/3 500和1/30 000。1/3由新生突变引起[1]。近年对DMD/BMD的研究取得了新的进展,致病基因已定位于Xp21,基因长2 300kb,含70个外显子,编码一种位于肌细胞膜上的骨架蛋白-“肌营养不良蛋白”(dystrophin),基因缺失或重复均导致进行性肌营养不良症[2,3]。Chamberlain等[4]采用9对引物多重聚合酶链反应(mPCR)检出DMD/BMD基因缺失的80%,Beggs等[5]设计了另外9对引物并联合应用这两组引物检出基因缺失的98%,从而对本病基因检测建立了一种简便快速的诊断方法。本组参照上述引物序列合成18对引物,对我院经临床,肌电图,血清酶学和肌活检确诊的96例无亲缘关系的DMD/BMD进行基因缺失筛查,并对我国DMD/BMD基因缺失的分布及其与临床表型的关系进行初步探讨。
, 百拇医药
对象和方法
一、对象
96例患者均为我院1992~1996年住院或门诊患者,均为男性,发病年龄6~18岁(平均12.4±3.3岁),根据丧失行走能力的年龄将患者分为4型:(1)Duchenne型(DMD)65例(68%),8~13岁以前丧失行走能力;(2)中间型6例(6%),13~16岁丧失行走能力;(3)Becker型(BMD)20例(21%),16岁后仍能行走;(4)5例(5%)8岁以下患者未能分型。临床均表现为四肢近端对称性进行性加重的肌肉萎缩,70%伴有腓肠肌或二头肌假肥大,均有Gower征阳性。肌电图检查显示典型肌源性改变,血清肌酸磷酸激酶(CK)及其同功酶MCK均显著增高,肌活检显示镶嵌分布的肌纤维萎缩,肥大,玻璃样变和坏死等典型病理改变。30例正常对照均为非神经肌病患者。
二、测定方法
1.DNA的提取:抽取患者和正常对照外周血5 ml,加入0.2%依地酸,乙二胺四乙酸(EDTA)抗凝,采用等渗溶血法裂解红细胞后离心分离白细胞,加入1%十二烷基硫酸钠(SDS)和蛋白酶K,37℃消化4~6小时,再加入1/3体积的饱和氯化钠,离心除去沉淀,用等体积的氯仿抽提两次,2.5倍体积的无水乙醇沉淀,75%乙醇漂洗,晾干后溶于TE(10 mmol/L Tris.CI, 1 mmol/L EDTA,pH 7.6),-20℃保存。
, 百拇医药
2.mPCR:18对引物参照Chamberlain和Beggs等[4,5]提供的序列由美国CyberSyn公司合成,经寡核苷酸纯化柱(OPC)纯化。将18对引物分为两组(第1组:外显子4,8,12,17,19,44,45,48,51;第2组:启动子,外显子3,6,13,43,47,50,52,60),分两步对全组患者进行mPCR扩增。50 μl PCR反应体系含670 mmol Tris-HCl(pH 8.0),166mmol(NH4)2SO4,67 mmol MgCl2,10 mmol β-巯基乙醇,10 μg/μl小牛血清白蛋白(BSA),25 mmol dNTP,每对引物各0.25 μmol,模板DNA 500 ng。将反应液97℃予变性10分钟,加入5UTaqDNA聚合酶(德国BM产品),混匀后加入35 μl石蜡油。按照94℃变性30秒,56℃复性30秒,70℃延伸2分钟进行30次循环,最后70℃延伸7分钟。取10 μl扩增产物用2%琼脂糖或6%聚丙烯酰胺凝胶在0.5×TBE中电泳,溴化已锭染色,在紫外灯下观察结果并摄影,或用0.2%硝酸银染色保留结果。
, http://www.100md.com
结果
96例患者用18对引物mPCR扩增共检出60例缺失,其中用第1组引物检出49例缺失,检出率为51%,用第2组引物在未检出缺失的47例中检出11例缺失,检出率为12%(图1,2)。两组引物检出的总缺失率为63%。60例缺失中DMD42例(70%),中间型3例(5%),BMD13例(22%),未分类2例(3%);41例为移码缺失,19例为整码缺失。全部缺失均经全长cDNA探针(1-2,2b-3,4-5a,5b-7,8,9-14)(质粒由Kunkel教授赠送)Southern杂交证实,用18对引物检出的缺失占全长cDNA探针检出缺失的97%。因两组引物均未覆盖基因中心区,用cDNA探针4-5a和5b-7在这一区域内检出的两例缺失漏检,漏检率为3%。本组用外显子60未检出缺失,可能与病例数较少有关。两步mPCR检出基因3′侧外显子44~52缺失43例,占全部缺失的72%,5′侧外显子1~19缺失17例,占28%。30例正常对照均未见基因缺失。
, http://www.100md.com
1:扩增带大小标志(PBR322HinfI);2:正常对照;3~7:DMD/BMD基因内缺失图1 第1组引物mPCR扩增DNA分析
1:正常对照;2~7:DMD/BMD不同基因内缺失
图2 第2组引物mPCR扩增DNA分析
讨论
一、应用mPCR技术
mPCR技术的应用对于DNA分析是一种革命性的探索。以往DMD/BMD基因缺失需要使用七个不同的亚克隆cDNA探针检测,基因组DNA至少需用两种不同的限制性内切酶消化,检测步骤极其繁琐。应用mPCR技术大大检化了Southern印迹分析,根据大部分缺失集中在两个区域的理论,仅检测70个外显子中的一部分即可检测出大部分缺失。Chamberlain等[4]首先在基因3′侧和5′侧以9个缺失热点外显子(4,8,12,17,19,44,45,48,51)的旁侧序列设计了9对引物,一次mPCR扩增即可检出用全长cDNA探针检出缺失的80%。
, 百拇医药
二、应用引物扩增
Beggs等[5]在mPCR研究的基础上选择另外8个缺失热点外显子(3,6,13,43,47,50,52,60)和启动子设计了相应的9对引物,进一步扩大了缺失的检测范围,使本病基因缺失检出率达到98%以上。上述两组引物联合应用不仅能扩增外显子44~52缺失热区内的大部分外显子,而且还能检测5′端和基因远端的缺失,并可分析突变对翻译阅读框架的影响。
三、与Southern印迹分析比较
本组应用18对引物对96例DMD/BMD进行两步mPCR扩增结果与Southern印迹分析的结果一致。第1组引物检出49例缺失,占全长cDNA探针检出缺失的79%,第2组引物检出11例缺失,占全长cDNA探针检出缺失的18%,两组引物检出缺失的总和占Southern印迹分析检出缺失的97%。这一结果与国外文献报道的缺失检出率接近[5,6]。实践证明这两组引物也适用于中国人DMD的基因诊断。应该指出的是,应用mPCR可以避免Southern杂交在基因诊断中的某些失误,例如两例应用cDNA8探针杂交显示缺失外显子48~52,但是用mPCR扩增时外显子51和52扩增带均显示清晰。Southern杂交出现失误的原因可能是由于DNA印迹转移不良,或在膜杂交的过程中出现气泡阻碍杂交过程。由于两套引物均未覆盖基因中心区,本组两例相当cDNA探针4-5a和5b-7检测区域内外显子20~40和21~42缺失漏检,漏检率仅为3%。但由于mPCR快速,准确,基因组DNA用量少,采用两套引物即可覆盖基因3′和5′侧大部分缺失热区外显子和基因的远侧端,其优越性已大大超过Southern印迹分析。mPCR对反应条件要求较高,使用国产Taq酶有时出现假阳性。改用BM(德国)Taq酶,在缓冲液中加入0.01%明胶,并将循环次数增加至30~35次,可稳获9条扩增带。还应避免基因组DNA保存时间过长,以免DNA发生降解而出现假阳性。
, 百拇医药
四、DMD/BMD基因缺失分布规律
本组资料分析显示,DMD/BMD基因缺失的分布确有一定的规律。60例缺失中43例(72%)分布于外显子44~52,相当于cDNA探针8和7的3′侧4个外显子覆盖的区域内。基因缺失范围的大小不尽相同,其中以外显子48,50,51和52受累最多见。17例(28%)缺失分布于5′侧外显子1~19,其中以外显子12~19受累最多见,1~9次之。这种缺失分布不均一的现象表明DMD/BMD的缺失热区主要分布在基因中心区3′侧外显子44~52或48~52,另一个缺失热区分布在基因5′侧外显子12~19。Koenig等[7]通过对329例DMD/BMD Southern印迹分析,发现其中75%的基因缺失分布在基因中心区3′侧,25%分布在基因5′侧,并将这两个区域称为缺失热区。这一基因缺失分布规律相继被以后的研究所证实[5,6,8]。由此可见,我国DMD/BMD基因缺失的分布规律与欧洲白种人基本相似。
, 百拇医药
五、DMD/BMD的临床表型与基因缺失类型的关系
近年研究证实,DMD/BMD的临床表型与基因缺失类型有密切的关系[7]。本组41例患者为移码缺失,因缺失破坏了翻译阅读框架而导致基因功能丧失,临床表型为严重的DMD,常于9~13岁丧失行走能力。19例为整码缺失,其中11例缺失位于外显子2~43,因缺失部位不是基因的关键部位,部分基因功能得以保留,临床表型为症状较轻的中间型和BMD,分别于13~16岁和16岁以后丧失行走能力;8例缺失位于基因中心区3′侧,缺失均累及外显子51,其中6例(75%)表现为严重的DMD,仅两例(25%)为轻型BMD。Koenig等[9]发现51号外显子恰与肌细胞膜骨架蛋白dystrophin绞链3相对应,绞链3对维持dystrophin的弹性和柔韧性起着重要作用。故累及基因中心区3′侧外显子51的整码缺失临床多为严重的DMD表型。
参考文献
, 百拇医药
1 Koenig M, Hoffman EP, Bertelson CJ, et al. Complete cloning of the Duchenne muscular dystrophy cDNA. Cell, 1987, 50:509-517.
2 Monaco AP, Bertelson CJ, Liechti-Callati S, et al. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 1988, 2:90-95.
3 Burmeister M, Monaco AP, Gillard EF, et al. A 10-megabase physical map of human Xp21, including the Duchenne muscular dystrophy gene. Genomics, 1988, 2:189-202.
, http://www.100md.com
4 Chamberlain JS, Gibbs RA, Ranier HE, et al. Multiplex PCR for the diagnosis of Duchenne muscular dystrophy. New York: Acade Press, 1990. 272-281.
5 Beggs AH, Koenig M, Frederick M, et al. Detection of 98% of DMD/BMD gene deletions by polymerase chain reaction. Hum Genet, 1990, 86:45-48.
6 Abbs S, Yau SC, Clark S, et al. A convenient multiplex PCR system for the detection of dystrophin gene deletions. J Med Genet, 1991, 28:304-311.
, 百拇医药
7 Koenig M, Beggs AH, Moyer M, et al. The molecular basis for Duchenne versus Becker muscular dystrophy: correlation of severity with type of deletion. Am J Hum Gennet, 1989, 45:489-506.
8 Patino A, Narbona J, Garcia-Delgado M. Molecular analysis of the Duchenne muscular dystrophy gene in Spanish individuals. Am J Med Genet, 1995, 59:182-187.
9 Koenig M, Kunkel LM. Detailed analysis of the repeat domain of dystrophin reveals four potential hinge segments that may confer flecibility. J Bio Chem, 1990, 256:4560-4566., 百拇医药