高效液相色谱测定司帕沙星胶囊含量方法的改进
作者:殷强
单位:常州市药品检验所,常州 213003
关键词:高效液相色谱法;司帕沙星;氧氟沙星
中国新药杂志001114 [摘要]目的:在3种ODS柱上对司帕沙星胶囊进行高效液相色谱法的色谱行为测试。方法:采用RP-HPLC法,以V(0.25%磷酸,用三乙胺调pH至3.0)∶V(乙腈)=70∶30为流动相,以氧氟沙星作为内标,检测波长298nm,在9.9~79.2μg/ml范围内线性关系良好(r=0.9995)。结果:该法简便、快速、准确、重现性好。结论:此法有利于从宏观上把握喹诺酮类药物的色谱行为规律,减少不必要的实验条件和次数,对ODS色谱柱的选择和分离度的改善均有指导意义。
[中图分类号]R927.2;R978 [文献标识码]B
[文章编号]1003-3734(2000)11-0772-02
, 百拇医药
Improvement in HPLC determination of sparfloxacin in capsules
YIN Qiang
(Changzhou Institute for Drug Control,Changzhou 213003,China)
[Abstract]Objective:To study the chromatographic behaviours of sparfloxacin in capsules on 3 ODS columns.Methods:The chromatographic behaviours of sparfloxacin in capsules were studied on 3 ODS columns.A mobile phase consisted of acetonitrile∶0.25% phosphoric acid (pH was adjusted to 3.0 by triethylamine)=30∶70 by volume,an UV detector at 298 nm and an internal standard,ofloxacin,were adotped.Results:An accurate and reproducible results with a linear range of 9.9~79.2μg/ml(r=0.9995) were obtained.Conclusion:The study is helpful in HPLC determination of quinolones in the following aspects:to recognize chromatographic behaviour,to optimize chromatographic conditions and to select internal standards.
, 百拇医药
[Key words]HPLC;sparfloxacin;ofloxacin
司帕沙星胶囊为新型喹诺酮类广谱抗菌药,对呼吸系统、肠道、胆道、泌尿生殖系统感染等具有很好的疗效。卫生部试行标准收载了该药的检测方法(简称原法),但实验中发现该法色谱图主峰在一般的ODS柱上拖尾严重,按外标法计算,结果误差较大。经实验摸索,改变了流动相缓冲液的成分,以氧氟沙星作为内标,检测并选出最佳分离条件,使主峰与内标峰达到基线分离,消除主峰与内标峰的拖尾,结果令人满意,现介绍如下。
1 仪器、药品和试剂
岛津LC-10AD高效液相色谱仪,SPD-10A紫外检测器,C-R6A记录仪。
司帕沙星对照品(含量98.80%)及其胶囊(由浙江海正药业股份有限公司提供,批号:980602,990301),氧氟沙星对照品由江苏省常州市聚荣药业股份有限公司提供,乙腈为色谱纯,三乙胺等试剂均为分析纯。
, 百拇医药
2 方法与结果
2.1 色谱条件 色谱柱:ODS-A 120A(6.0mm×150mm,5μm,日本YMC公司)。流动相:V(体积分数为0.25%的磷酸溶液,用三乙胺调pH至3.0)∶V(乙腈)=70∶30,流速1.5ml/min。检测波长298nm,柱温30℃,灵敏度0.05AUFS。在上述色谱条件下,主峰与内标峰得到有效的分离,分离度为3.8,见图1。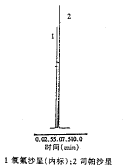
1 氧氟沙星(内标);2 司帕沙星
图1 司帕沙星HPLC色谱图
2.2 标准溶液的准备 对照品溶液:精密称取司帕沙星对照品适量,置于25 ml量瓶中,用乙腈溶解并稀释制成0.4mg/ml的溶液。内标溶液:精确称取氧氟沙星对照品适量,置于25ml量瓶中,加乙腈溶解并稀释制成0.4mg/ml的溶液。
, http://www.100md.com
2.3 线性关系考察 精密量取对照品溶液0.25,0.50,1.0,1.5,2.0ml分别置于10ml量瓶中,各精密加内标溶液0.50ml,用流动相稀释至刻度,摇匀,分别进样20μl,进行色谱分析,经直线线性回归,得回归方程为:Y=0.03797X-0.02078, r=0.9995。
结果表明,司帕沙星浓度在9.9~79.2μg/ml范围内,呈良好的线性关系。
2.4 精密度测定 分别精密量取前项下的对照品溶液0.50ml与内标溶液0.50ml,置同一10ml量瓶中,用流动相稀释至刻度,摇匀,进样20μl,连续进样8次,测得校正因子为1.2421,RSD为0.4%,其精密度良好。
2.5 加样回收率 向已知含量的样品加入一定量对照品,依法测定其含量,重复5个样品测定,司帕沙星回收率为98.9%,RSD为0.64%。
, 百拇医药 2.6 样品测定 取本品20粒,去除囊壳,内容物混匀,精称适量(约相当于司帕沙星25mg)置50ml量瓶中,加乙腈溶解并稀释至刻度。精密量取该溶液0.50ml与内标溶液0.50ml,置同一10ml量瓶中,用流动相稀释至刻度,摇匀,0.45μm滤膜过滤,进样20μl测定,结果见表1。
表1 样品测定结果(n=4) 批 号
含量(标示量)
RSD(%)
980602
100.6
0.82
990301
96.3
, 百拇医药
1.00
3 讨论
3.1 流动相的选择 原法流动相中采用磷酸盐缓冲液,本实验采用不含盐类的缓冲液,不但简化了操作步骤,而且对色谱柱也起到了保护作用。由于司帕沙星和氧氟沙星均为碱性药物,在流动相中加入扫尾剂三乙胺,可抑制或掩蔽固定相表面的游离硅醇基的活性,使碱性药物易于出峰,且峰形较好,就多数碱性药物来说,选用pH为3.0的缓冲液较为适宜,若pH值再升高则碱性药物在柱上的保留时间将延长,增加了分析所耗的时间[1]。在实验中,我们把缓冲液与乙腈的体积之比分别调至85∶15,80∶20,75∶25及70∶30。结果表明,随着乙腈浓度的增大,主峰与内标峰之间分离情况逐渐改善,至70∶30时,分离良好,主峰与内标峰无拖尾现象。故认为V(体积分数为0.25%的磷酸-三乙胺缓冲液,pH为3.0)∶V(乙腈)=70∶30为优化流动相。
3.2 色谱柱的选择 在实验中,我们发现不同牌号的色谱柱对本品测定结果影响很大。在美国惠普公司的ZORBAX ODS C18(4.6mm×150mm,5μm)柱上,无论改变流速、pH值及乙腈与缓冲液的体积比,均无法改变主峰与内标峰严重拖尾现象。这是由于普通柱中,固定相表面的游离硅醇基对碱性药物的色谱行为产生了严重的影响。为此,我们选用了美国惠普公司的ZORBAX SB-C18(4.6mm×150mm,5μm)色谱柱和日本YMC-pack,ODS-A,120A(6.0mm×150mm,5μm)色谱柱,它们均采用特殊的基团封端技术,避免未反应的硅醇基与碱性物质相互作用,引起峰拖尾。司帕沙星与氧氟沙星在3种ODS色谱柱上的分离性能见表2。
, 百拇医药
表2 3种ODS色谱柱的分离性能 优化流动相
分离度
以司帕沙星峰计
保留时间(min)
理论塔板数
拖尾因子
司帕沙星峰
氧氟沙星峰
日本YMC ODS
3.8
10 823
1.0
, 百拇医药
4.5
3.1
美国HP ZORBAX
3.0
9 313
1.0
2.1
1.3
ODS SB-C18
美国HP ZORBAX
1.1
1 654
, 百拇医药
6.2
2.1
1.4
ODS C18
原法流动相
8
4 080
1.0
9.5
4.9
(日本YMC ODS)
3.3 内标物质的选择[2]分别选用了甲硝唑、西咪替丁、氧氟沙星和诺氟沙星作为内标,从色谱峰保留时间及与主峰的分离度性能来看,最终选用氧氟沙星作为内标。上述色谱条件,同样也适用于氧氟沙星及其制剂及其他喹诺酮类药物含量测定的优化。
, 百拇医药
[作者简介]殷强(1966-),男,副主任药师,主要从事高效液相色谱分析。
[参考文献]
[1]李修禄.用HPLC法分析碱性含氮有机药物的进展[J].国外医学药学分册,1991,18(5)∶265-269.
[2]梁贵键,朱莉,邹凤君,等.根据色谱保留值与流动相pH值的函数关系对212种常用药物的分类[J].色谱,1996,14(3)∶196-198.
收稿日期:2000-02-21
修回日期:2000-08-16, http://www.100md.com
单位:常州市药品检验所,常州 213003
关键词:高效液相色谱法;司帕沙星;氧氟沙星
中国新药杂志001114 [摘要]目的:在3种ODS柱上对司帕沙星胶囊进行高效液相色谱法的色谱行为测试。方法:采用RP-HPLC法,以V(0.25%磷酸,用三乙胺调pH至3.0)∶V(乙腈)=70∶30为流动相,以氧氟沙星作为内标,检测波长298nm,在9.9~79.2μg/ml范围内线性关系良好(r=0.9995)。结果:该法简便、快速、准确、重现性好。结论:此法有利于从宏观上把握喹诺酮类药物的色谱行为规律,减少不必要的实验条件和次数,对ODS色谱柱的选择和分离度的改善均有指导意义。
[中图分类号]R927.2;R978 [文献标识码]B
[文章编号]1003-3734(2000)11-0772-02
, 百拇医药
Improvement in HPLC determination of sparfloxacin in capsules
YIN Qiang
(Changzhou Institute for Drug Control,Changzhou 213003,China)
[Abstract]Objective:To study the chromatographic behaviours of sparfloxacin in capsules on 3 ODS columns.Methods:The chromatographic behaviours of sparfloxacin in capsules were studied on 3 ODS columns.A mobile phase consisted of acetonitrile∶0.25% phosphoric acid (pH was adjusted to 3.0 by triethylamine)=30∶70 by volume,an UV detector at 298 nm and an internal standard,ofloxacin,were adotped.Results:An accurate and reproducible results with a linear range of 9.9~79.2μg/ml(r=0.9995) were obtained.Conclusion:The study is helpful in HPLC determination of quinolones in the following aspects:to recognize chromatographic behaviour,to optimize chromatographic conditions and to select internal standards.
, 百拇医药
[Key words]HPLC;sparfloxacin;ofloxacin
司帕沙星胶囊为新型喹诺酮类广谱抗菌药,对呼吸系统、肠道、胆道、泌尿生殖系统感染等具有很好的疗效。卫生部试行标准收载了该药的检测方法(简称原法),但实验中发现该法色谱图主峰在一般的ODS柱上拖尾严重,按外标法计算,结果误差较大。经实验摸索,改变了流动相缓冲液的成分,以氧氟沙星作为内标,检测并选出最佳分离条件,使主峰与内标峰达到基线分离,消除主峰与内标峰的拖尾,结果令人满意,现介绍如下。
1 仪器、药品和试剂
岛津LC-10AD高效液相色谱仪,SPD-10A紫外检测器,C-R6A记录仪。
司帕沙星对照品(含量98.80%)及其胶囊(由浙江海正药业股份有限公司提供,批号:980602,990301),氧氟沙星对照品由江苏省常州市聚荣药业股份有限公司提供,乙腈为色谱纯,三乙胺等试剂均为分析纯。
, 百拇医药
2 方法与结果
2.1 色谱条件 色谱柱:ODS-A 120A(6.0mm×150mm,5μm,日本YMC公司)。流动相:V(体积分数为0.25%的磷酸溶液,用三乙胺调pH至3.0)∶V(乙腈)=70∶30,流速1.5ml/min。检测波长298nm,柱温30℃,灵敏度0.05AUFS。在上述色谱条件下,主峰与内标峰得到有效的分离,分离度为3.8,见图1。
1 氧氟沙星(内标);2 司帕沙星
图1 司帕沙星HPLC色谱图
2.2 标准溶液的准备 对照品溶液:精密称取司帕沙星对照品适量,置于25 ml量瓶中,用乙腈溶解并稀释制成0.4mg/ml的溶液。内标溶液:精确称取氧氟沙星对照品适量,置于25ml量瓶中,加乙腈溶解并稀释制成0.4mg/ml的溶液。
, http://www.100md.com
2.3 线性关系考察 精密量取对照品溶液0.25,0.50,1.0,1.5,2.0ml分别置于10ml量瓶中,各精密加内标溶液0.50ml,用流动相稀释至刻度,摇匀,分别进样20μl,进行色谱分析,经直线线性回归,得回归方程为:Y=0.03797X-0.02078, r=0.9995。
结果表明,司帕沙星浓度在9.9~79.2μg/ml范围内,呈良好的线性关系。
2.4 精密度测定 分别精密量取前项下的对照品溶液0.50ml与内标溶液0.50ml,置同一10ml量瓶中,用流动相稀释至刻度,摇匀,进样20μl,连续进样8次,测得校正因子为1.2421,RSD为0.4%,其精密度良好。
2.5 加样回收率 向已知含量的样品加入一定量对照品,依法测定其含量,重复5个样品测定,司帕沙星回收率为98.9%,RSD为0.64%。
, 百拇医药 2.6 样品测定 取本品20粒,去除囊壳,内容物混匀,精称适量(约相当于司帕沙星25mg)置50ml量瓶中,加乙腈溶解并稀释至刻度。精密量取该溶液0.50ml与内标溶液0.50ml,置同一10ml量瓶中,用流动相稀释至刻度,摇匀,0.45μm滤膜过滤,进样20μl测定,结果见表1。
表1 样品测定结果(n=4) 批 号
含量(标示量)
RSD(%)
980602
100.6
0.82
990301
96.3
, 百拇医药
1.00
3 讨论
3.1 流动相的选择 原法流动相中采用磷酸盐缓冲液,本实验采用不含盐类的缓冲液,不但简化了操作步骤,而且对色谱柱也起到了保护作用。由于司帕沙星和氧氟沙星均为碱性药物,在流动相中加入扫尾剂三乙胺,可抑制或掩蔽固定相表面的游离硅醇基的活性,使碱性药物易于出峰,且峰形较好,就多数碱性药物来说,选用pH为3.0的缓冲液较为适宜,若pH值再升高则碱性药物在柱上的保留时间将延长,增加了分析所耗的时间[1]。在实验中,我们把缓冲液与乙腈的体积之比分别调至85∶15,80∶20,75∶25及70∶30。结果表明,随着乙腈浓度的增大,主峰与内标峰之间分离情况逐渐改善,至70∶30时,分离良好,主峰与内标峰无拖尾现象。故认为V(体积分数为0.25%的磷酸-三乙胺缓冲液,pH为3.0)∶V(乙腈)=70∶30为优化流动相。
3.2 色谱柱的选择 在实验中,我们发现不同牌号的色谱柱对本品测定结果影响很大。在美国惠普公司的ZORBAX ODS C18(4.6mm×150mm,5μm)柱上,无论改变流速、pH值及乙腈与缓冲液的体积比,均无法改变主峰与内标峰严重拖尾现象。这是由于普通柱中,固定相表面的游离硅醇基对碱性药物的色谱行为产生了严重的影响。为此,我们选用了美国惠普公司的ZORBAX SB-C18(4.6mm×150mm,5μm)色谱柱和日本YMC-pack,ODS-A,120A(6.0mm×150mm,5μm)色谱柱,它们均采用特殊的基团封端技术,避免未反应的硅醇基与碱性物质相互作用,引起峰拖尾。司帕沙星与氧氟沙星在3种ODS色谱柱上的分离性能见表2。
, 百拇医药
表2 3种ODS色谱柱的分离性能 优化流动相
分离度
以司帕沙星峰计
保留时间(min)
理论塔板数
拖尾因子
司帕沙星峰
氧氟沙星峰
日本YMC ODS
3.8
10 823
1.0
, 百拇医药
4.5
3.1
美国HP ZORBAX
3.0
9 313
1.0
2.1
1.3
ODS SB-C18
美国HP ZORBAX
1.1
1 654
, 百拇医药
6.2
2.1
1.4
ODS C18
原法流动相
8
4 080
1.0
9.5
4.9
(日本YMC ODS)
3.3 内标物质的选择[2]分别选用了甲硝唑、西咪替丁、氧氟沙星和诺氟沙星作为内标,从色谱峰保留时间及与主峰的分离度性能来看,最终选用氧氟沙星作为内标。上述色谱条件,同样也适用于氧氟沙星及其制剂及其他喹诺酮类药物含量测定的优化。
, 百拇医药
[作者简介]殷强(1966-),男,副主任药师,主要从事高效液相色谱分析。
[参考文献]
[1]李修禄.用HPLC法分析碱性含氮有机药物的进展[J].国外医学药学分册,1991,18(5)∶265-269.
[2]梁贵键,朱莉,邹凤君,等.根据色谱保留值与流动相pH值的函数关系对212种常用药物的分类[J].色谱,1996,14(3)∶196-198.
收稿日期:2000-02-21
修回日期:2000-08-16, http://www.100md.com