网状色素性皮病一例
作者:肖毅 赵玉铭 翟宁 耿龙 宋芳吉 陈洪铎 蔡昌盛
单位:110001 沈阳,中国医科大学第一临床学院皮肤科 (肖毅 赵玉铭 翟宁 耿龙 宋芳吉 陈洪铎);辽宁中医学院附属医院皮肤科(蔡昌盛)
关键词:
中国皮肤科杂志980347 网状色素性皮病(dermatopathia pigmentosa reticularis, DPR)是一种罕见的、临床上主要表现为网状色素沉着斑、非瘢痕性脱发及甲营养不良的皮肤病。1958年Ha uss和Oberste-Lehn首次报道了姊妹同患DPR后,迄今共报道12例,除一例为亚洲人外[1],均为欧美所报告,国内尚未见报道。本例为有典型DPR症状的14岁汉族女性,伴有视力改变、牙龈异常及粟丘疹样皮损。我们还对该患及其近亲进行了HLA单倍型分析。
患者女,14岁,汉族。出生后发现颜面、躯干、四肢皮肤黑褐色斑,随年龄增长色斑颜色加深。近6年来手足皮肤粗糙增厚,头发及眉毛稀疏脱落。无明显多汗及少汗,手足背未出现过水疱。无关节痛及出血倾向。患者父母否认家族中有类似病史。体检:一般情况好,发育中 等,智力正常。心肺无异常,肝脾未触及。双眼近视(-2.5D)伴散光、斜视,角膜正常,结膜无色素沉着。唇粘膜角化增厚,见棘刺状斑块。右门牙及尖牙近牙冠1/2处牙釉质呈淡褐色,牙乳头充血、水肿,口腔粘膜无色素斑。双耳听力正常。皮肤科检查:躯干、四肢棕褐色网状色素沉着斑(图1),躯干部还可见一些略凹陷的色素减退斑;颜面及双前臂散在粟粒大小乳白色粟丘疹样皮损(图2);掌跖斑状角化,指纹存在,部分指间关节伸侧皮肤呈指垫样角化肥厚;乳头色素明显增多,乳晕及外阴部皮肤肥厚,皮嵴增高,皮沟加深,有黑褐色色素沉着;指甲正常,双侧趾甲变薄,光泽消失,见棕褐色横纹;额部毛发稀疏,发质干脆 ,双侧眉毛稀疏,外1/3脱落,腋毛及阴毛缺如。实验室检查:血、尿常规及肝功能正常, 胸部X线检查未见异常。组织病理:表皮变薄,基底层色素增多,有灶性
, 百拇医药
图1 患者躯干部网状色素沉着斑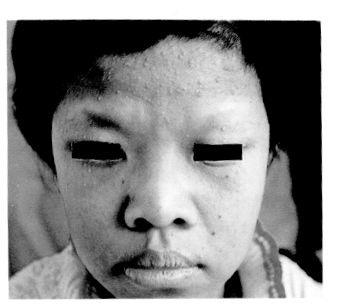
图2 患者颜面部粟丘疹样皮损
基底细胞液化变性,真皮浅层少许色素滴落。家系HLA单倍型分析如下:父:A9-B22 /A2-B46;母:A2-B48/A30+31-B46;患者:A2-B48/A3-B15;胞弟:A2-B48/A9-B22 。
讨论 DPR的主要诊断依据为网状色素沉着斑、非瘢痕性脱发及甲营养不良三联征。本例DPR还伴有掌跖斑状角化、眉毛稀疏脱落、腋毛及阴毛缺如、乳头色素增多等次要表现,与文献报道一致[1~3]。此外,还见到以往未报道的表现,如近视、散光、斜视、粟丘疹样皮损、唇粘膜角化增厚及棘刺状斑、牙乳头充血与水肿 、个别指间关节伸侧呈指垫样角化肥厚。这些表现是否与DPR有关,尚需进一步观察。
, 百拇医药
DPR中还可见到少汗或多汗、手足背非瘢痕性水疱、无指纹等,但本例未见到。文献报道一些DPR病例尚有角膜点状斑、鱼鳞病样改变、手足肘膝皮肤萎缩、结膜及口腔粘膜色素斑、 网状色素斑的光敏反应、背部毛周角化、皮肤神经纤维瘤、龋齿、癫痫、尿道口狭小、单侧 睾丸萎缩和单纯性甲状腺肿等[1~3]。
本病病因不明。1992年Heimer[3]等报道了一个五代9位患者(其中2人被医生证实)的DPR家系,认为DPR为常染色体显性遗传。其他亦有报道姊妹、姐弟及表兄或叔伯同患的病例[2]。本例HLA分型示A2-B48/A3-B15,与患者父亲的单倍型不同(A9-B22/A2-B46),但未能肯定是否为其生父,无法进行家系研究 ,故难以确定该例为遗传抑或散发。
DPR需与其他遗传或先天性色素异常性皮肤病鉴别[1]:进行性肢端色素沉着病(acromelanosis progressiva)、遗传性对称性色素异常症(hereditary sym metrical dyschromatosis of Dohi)、网状肢端色素沉着症(reticular acropigmentation of Kitamura)、肢体异色症(heterochromia extremitarum)及屈侧网状色素性皮病(reticul ate pigmented dermatosis of the flexures)的皮疹分布于肢体或肢端,其特点亦与本病 不同,可资鉴别。先天性弥漫性色素斑(congenital diffuse mottling of the skin)与DPR 的鉴别要点是依据其病理学特点和其他伴随症状。遗传性泛发性色素异常症(hereditary un iversal dyschromatosis)的色素减退斑散在分布于色素增加斑之间,可伴身材矮小及高音区耳聋。先天性角化不良综合征(dyskeratosis congenita syndrome)为性联隐性遗传性疾病,几乎均发生于男性,以5岁后发病为特征,常伴有粘膜白斑和血恶液质等。弗-雅综合征(Franceschetti-Jadassohn syndrome或Nae-
, http://www.100md.com
geli syndrome)是血小板功能异常 所致的常 染色体显性遗传性疾病,常有皮肤粘膜等出血,皮肤色素沉着范围不超出躯干及颈部,呈污灰色,随年龄增长而减轻,伴牙釉质缺损,常致早期失去全部牙齿。本例根据临床上特异的三联征及其它表现可确诊为DPR,而除外其他先天性色素异常性皮肤病。
参考文献
1 Bu TS, Kim YK, Whang KU. A case of dermatopathia pigment osa reticularis. J Dermatol, 1997,24∶266.
2 Maso MJ, Schwartz RA, Lambert WC. Dermatopathia pigmentosa reticularis. Arch Dermatol, 1990,126∶935.
3 Heimer WJ Ⅱ, Brauner G, James WD. Dermatopathia pigmentosa reticularis : a repor t of a family demonstrating autosomal dominant inheritance. J Am Acad Dermatol, 1992,26(2 pt 2)∶298., 百拇医药
单位:110001 沈阳,中国医科大学第一临床学院皮肤科 (肖毅 赵玉铭 翟宁 耿龙 宋芳吉 陈洪铎);辽宁中医学院附属医院皮肤科(蔡昌盛)
关键词:
中国皮肤科杂志980347 网状色素性皮病(dermatopathia pigmentosa reticularis, DPR)是一种罕见的、临床上主要表现为网状色素沉着斑、非瘢痕性脱发及甲营养不良的皮肤病。1958年Ha uss和Oberste-Lehn首次报道了姊妹同患DPR后,迄今共报道12例,除一例为亚洲人外[1],均为欧美所报告,国内尚未见报道。本例为有典型DPR症状的14岁汉族女性,伴有视力改变、牙龈异常及粟丘疹样皮损。我们还对该患及其近亲进行了HLA单倍型分析。
患者女,14岁,汉族。出生后发现颜面、躯干、四肢皮肤黑褐色斑,随年龄增长色斑颜色加深。近6年来手足皮肤粗糙增厚,头发及眉毛稀疏脱落。无明显多汗及少汗,手足背未出现过水疱。无关节痛及出血倾向。患者父母否认家族中有类似病史。体检:一般情况好,发育中 等,智力正常。心肺无异常,肝脾未触及。双眼近视(-2.5D)伴散光、斜视,角膜正常,结膜无色素沉着。唇粘膜角化增厚,见棘刺状斑块。右门牙及尖牙近牙冠1/2处牙釉质呈淡褐色,牙乳头充血、水肿,口腔粘膜无色素斑。双耳听力正常。皮肤科检查:躯干、四肢棕褐色网状色素沉着斑(图1),躯干部还可见一些略凹陷的色素减退斑;颜面及双前臂散在粟粒大小乳白色粟丘疹样皮损(图2);掌跖斑状角化,指纹存在,部分指间关节伸侧皮肤呈指垫样角化肥厚;乳头色素明显增多,乳晕及外阴部皮肤肥厚,皮嵴增高,皮沟加深,有黑褐色色素沉着;指甲正常,双侧趾甲变薄,光泽消失,见棕褐色横纹;额部毛发稀疏,发质干脆 ,双侧眉毛稀疏,外1/3脱落,腋毛及阴毛缺如。实验室检查:血、尿常规及肝功能正常, 胸部X线检查未见异常。组织病理:表皮变薄,基底层色素增多,有灶性
, 百拇医药
图1 患者躯干部网状色素沉着斑
图2 患者颜面部粟丘疹样皮损
基底细胞液化变性,真皮浅层少许色素滴落。家系HLA单倍型分析如下:父:A9-B22 /A2-B46;母:A2-B48/A30+31-B46;患者:A2-B48/A3-B15;胞弟:A2-B48/A9-B22 。
讨论 DPR的主要诊断依据为网状色素沉着斑、非瘢痕性脱发及甲营养不良三联征。本例DPR还伴有掌跖斑状角化、眉毛稀疏脱落、腋毛及阴毛缺如、乳头色素增多等次要表现,与文献报道一致[1~3]。此外,还见到以往未报道的表现,如近视、散光、斜视、粟丘疹样皮损、唇粘膜角化增厚及棘刺状斑、牙乳头充血与水肿 、个别指间关节伸侧呈指垫样角化肥厚。这些表现是否与DPR有关,尚需进一步观察。
, 百拇医药
DPR中还可见到少汗或多汗、手足背非瘢痕性水疱、无指纹等,但本例未见到。文献报道一些DPR病例尚有角膜点状斑、鱼鳞病样改变、手足肘膝皮肤萎缩、结膜及口腔粘膜色素斑、 网状色素斑的光敏反应、背部毛周角化、皮肤神经纤维瘤、龋齿、癫痫、尿道口狭小、单侧 睾丸萎缩和单纯性甲状腺肿等[1~3]。
本病病因不明。1992年Heimer[3]等报道了一个五代9位患者(其中2人被医生证实)的DPR家系,认为DPR为常染色体显性遗传。其他亦有报道姊妹、姐弟及表兄或叔伯同患的病例[2]。本例HLA分型示A2-B48/A3-B15,与患者父亲的单倍型不同(A9-B22/A2-B46),但未能肯定是否为其生父,无法进行家系研究 ,故难以确定该例为遗传抑或散发。
DPR需与其他遗传或先天性色素异常性皮肤病鉴别[1]:进行性肢端色素沉着病(acromelanosis progressiva)、遗传性对称性色素异常症(hereditary sym metrical dyschromatosis of Dohi)、网状肢端色素沉着症(reticular acropigmentation of Kitamura)、肢体异色症(heterochromia extremitarum)及屈侧网状色素性皮病(reticul ate pigmented dermatosis of the flexures)的皮疹分布于肢体或肢端,其特点亦与本病 不同,可资鉴别。先天性弥漫性色素斑(congenital diffuse mottling of the skin)与DPR 的鉴别要点是依据其病理学特点和其他伴随症状。遗传性泛发性色素异常症(hereditary un iversal dyschromatosis)的色素减退斑散在分布于色素增加斑之间,可伴身材矮小及高音区耳聋。先天性角化不良综合征(dyskeratosis congenita syndrome)为性联隐性遗传性疾病,几乎均发生于男性,以5岁后发病为特征,常伴有粘膜白斑和血恶液质等。弗-雅综合征(Franceschetti-Jadassohn syndrome或Nae-
, http://www.100md.com
geli syndrome)是血小板功能异常 所致的常 染色体显性遗传性疾病,常有皮肤粘膜等出血,皮肤色素沉着范围不超出躯干及颈部,呈污灰色,随年龄增长而减轻,伴牙釉质缺损,常致早期失去全部牙齿。本例根据临床上特异的三联征及其它表现可确诊为DPR,而除外其他先天性色素异常性皮肤病。
参考文献
1 Bu TS, Kim YK, Whang KU. A case of dermatopathia pigment osa reticularis. J Dermatol, 1997,24∶266.
2 Maso MJ, Schwartz RA, Lambert WC. Dermatopathia pigmentosa reticularis. Arch Dermatol, 1990,126∶935.
3 Heimer WJ Ⅱ, Brauner G, James WD. Dermatopathia pigmentosa reticularis : a repor t of a family demonstrating autosomal dominant inheritance. J Am Acad Dermatol, 1992,26(2 pt 2)∶298., 百拇医药