母系遗传性聋大家系听力学调查及线粒体DNA突变分析
作者:邢光前 卜行宽 严明 陆玲 杨舜英
单位:邢光前(210029 南京医科大学第一附属医院耳鼻咽喉科);卜行宽(210029 南京医科大学第一附属医院耳鼻咽喉科);陆玲(210029 南京医科大学第一附属医院耳鼻咽喉科);杨舜英(210029 南京医科大学第一附属医院耳鼻咽喉科);严明(南京铁道医学院分子生物学研究)
关键词:遗传性疾病;听觉丧失;感音神经性;DNA;线粒体;DNA突变分析;聚合酶链反应
中华耳鼻咽喉科杂志000206 【摘要】 目的 探讨母系遗传性非综合征性耳聋的听力学特征及分子遗传学机制。方法 选择一遗传聋大家系中4代41人为研究对象, 详细询问病史和全身体格检查, 行系统听力学测试和线粒体DNA(mtDNA)A1555G突变分析。结果 20例母系亲属均存在A1555G点突变;纯音测听17例呈程度不等的感音神经性聋,双耳对称,发病年龄1~50岁, 5例听力近11年间呈进行性下降;声导抗测试均为A型鼓室曲线,12耳可在低感觉级引出镫骨肌反射;听性脑干反应( auditory brainstem response,ABR)和瞬态诱发耳声发射测试无蜗后病变证据。21名父系亲属及配偶无听力障碍,mtDNA A1555G突变检测阴性。结论 本家系以迟发性进行性耳蜗性聋为特点,导致听力损失的内在因素为mtDNA A1555G突变,而环境因素和核基因可能也参与了突变体mtDNA表型表达的调节。
, http://www.100md.com
Audiological findings and mitochondrial DNA mutation in a large family with matrilineal sensorineural hearing loss
XING Guangqian, BU Xingkuan, YAN Ming, et al.
(Department of Otorhinolaryngology, First Affiliated Hospital, Nanjing Medical University, Nanjing 210029,China)
Corresponding author: XING Guangqian
【Abstract】 Objective To explore audiological features of matrilineal non-syndromic deafness and its molecular mechanism. Methods A large family with 41 members having inherited deafness was studied. Complete history and the data of general and otolaryngological examinations were collected. All subjects were screened for mitochondrial DNA A1555G mutation by molecular analysis. Audiological evaluation included pure-tone audiometry, auditory brainstem responses and transiently evoked otoacoustic emissions. Results All subjects were in good health generally. Molecular analysis showed that all maternal relatives with or without hearing loss harbored the A1555G mitochondrial mutation. No mutation was found among spouses and paternal relatives. Audiological results showed notable symmetric bilateral sensorineural hearing loss in 17 of 20 maternal relatives, in which 5 cases had a progressive hearing loss in the recent 11 years. The age of appearance of hearing loss ranged from 1 to 50 years. Conclusion All hearing-impaired subjects of this family had late-onset sensorineural hearing loss. Most of which were progressive. The A1555G mitochondrial mutation in the 12S rRNA gene is responsible for the disorder. Other factors, such as nuclear genes or environmental determinants, may influence the clinical expression of mutant mtDNA.
, 百拇医药
【Key words】 Hereditary diseases; Hearing loss,sensorineural; DNA, mitochondrial; DNA mutational analysis; Polymerase chain reaction
1987年我们在江苏省淮阴县发现一遗传聋大家系(6代507人),对其中4代386人进行了遗传学和听力学调查,确诊遗传聋患者137人[1]。为进一步探讨该家系耳聋发生的分子遗传学机制,于1998年3月对其中一分支家系的4代41人进行了系谱复核和系统听力学测试,系谱分析表明该家系耳聋患者呈典型的母系遗传方式传递。同时应用聚合酶链反应(polymerase chain reaction,PCR)产物酶切及PCR产物克隆序列测定技术,对目前文献报道的与非综合征性母系遗传聋相关的mtDNA突变位点――12S rRNA基因的A1555G[2]进行了研究,发现该家系中母系亲属均存在A1555G点突变,而家系配偶及男性个体的后代该突变检测阴性,提示mtDNA A1555G突变是该家系致聋的分子基础之一。
, http://www.100md.com
材料与方法
一、一般资料
该分支家系包括4代41人,其中直系亲属36人(母系亲属20人,父系亲属16人),配偶5人,遗传系谱见图1。母系亲属中原有15人于1987年曾接受过调查,调查内容包括纯音听阈测试和阈上功能检查、声导抗测试及听性脑干反应( auditory brainstem response,ABR)检测;另5人为近年来繁衍后代,属初查。对全体成员行详细病史询问和全身体格检查,尤其注意有无听力下降、眩晕、耳鸣及有无耳毒性药物和噪声接触史,如有听力损失,则详细记录发生年龄及进展情况。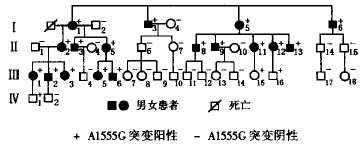
图1 耳聋核心家系图
二、mtDNA A1555G突变分析
, 百拇医药 1.DNA提取及模板mtDNA的扩增:全体成员各抽取外周静脉血5~10 ml,按酚/氯仿抽提法提取DNA。以mtDNA寡核苷酸(nt)1326~1335和(nt)1684~1704为引物,家系成员外周血白细胞粗提DNA为模板,扩增mtDNA (nt)1 326~1 704基因片段。PCR反应参数为:94℃ 4 min变性;94℃ 45 s,60℃ 45 s,72℃ 45 s,共35个循环。2%琼脂糖凝胶电泳观察PCR反应结果。
2.PCR产物纯化和酶切:使用宝灵曼公司PCR产物纯化试剂盒,按使用说明操作,纯化PCR产物。取经纯化的1555位点PCR产物10 μL,加入限制性内切酶Alw26 I (MBI公司,德国)10 U,37℃酶切1 h,2%琼脂糖凝胶电泳观察酶切结果。
3.PCR产物克隆和序列测定:取PCR纯化产物1 μL,T4 DNA连接酶缓冲液1 μL,PEGM-T Vector 1 μL, DNA连接酶1 μL, 双蒸水(ddH2O)6 μL,4℃连接24 h。常规法转化细菌DH5α,涂在含X-gal的LB固体培养基上,37℃培养。选取板上的白色菌落,重新划线培养。单菌落PCR检测,证实有插入片段的阳性克隆后,培养细菌,小制备碱法提取质粒。使用T7启动子引物,377全自动测序仪测序。
, http://www.100md.com
三、听力学检测
除1例2岁的母系亲属听阈由ABR测试结果确定外,全体成员行纯音听阈测试和声导抗测试。纯音听阈以0.5、1、2和4 kHz气导平均值计算,听力损失程度则根据WHO推荐草案[3]确定,即:轻度26~40 dB HL,中度41~60 dB HL,重度61~80 dB HL,极重度≥81 dB HL。进行性听力下降定义为:1987年和本次测试0.5~4 kHz纯音听阈平均提高≥10 dB或其间任意2个频率提高≥15 dB。
对5例初查的母系亲属同时行ABR和瞬态诱发性耳声发射(transiently evoked otoacoustic emission,TEOAE)检查。ABR测试用RION ER-02型诱发电位仪,Ⅴ波反应阈≤30 dB nHL定为正常标准。TEOAE测试用Celesta 503声发射仪进行,刺激声为短声,强度为80 dB peSPL,脉宽80 μs,采用非线性(3+1)给声方式,叠加1 000次,扫描20 ms,延迟5 ms记录,以波形总相关系数r≥0.6为正常标准。
, http://www.100md.com
所有检查在符合要求的隔声室或隔声电磁屏蔽室中进行,仪器均经校准合格使用。
结果
一、一般资料
母系亲属中15例诉听力下降, 均为1987年接受过调查者,根据病史及听力检测结果综合判断其发病年龄为1~50岁。其中10例主诉听力损失进行性加重,5例近11年来继续恶化;4例伴高音调耳鸣;2例有一过性眩晕发作;6例曾应用常规剂量氨基糖甙类抗生素(aminoglycoside antibiotics,AmAn),其中4例(Ⅰ6、Ⅱ11、Ⅲ1、Ⅲ2)在耳聋前3个月内用药,发病年龄1~3岁,另2例应用后原有听力损失急剧加重;2例2耳伴慢性化脓性中耳炎。父系亲属及配偶共21人,9人曾因各种原因接受过AmAn注射治疗。耳鼻咽喉及全身检查无异常。
二、mtDNA 1555位点突变检测结果
, 百拇医药
使用引物mtDNA (nt)1326-1335和(nt)1684-1704扩增家系中所有成员,均可见一379 bp扩增带。由于正常个体mtDNA 1555位点包含有限制性内切酶Alw26 I的切割识别位点GAGA1555C,酶切后可见153 bp和226 bp两条带,当存在A1555G突变时该酶切位点消失,酶切后仅见379 bp一条带。本家系成员酶切结果表明,20例母系亲属均存在mtDNA A1555G突变,而家系中配偶和男性个体(包括男性患者)的后代A1555G突变阴性(图2)。
图2 部分家系成员PCR产物酶切电泳图。M为标准物
对家系成员Ⅰ1、Ⅱ2、Ⅲ1、Ⅳ1 mtDNA 1555位点的测序结果表明,1555位点存在A→G点突变(H链,图3),而配偶和男性后代的1555位碱基为正常的A(L链)。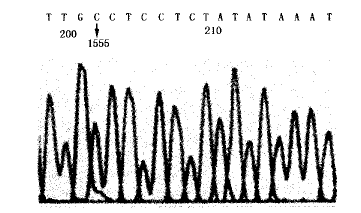
, 百拇医药
图3 mtDNA 1555位点测序结果(H链)
三、听力学检测结果
1.纯音测听:16名父系亲属和4名配偶纯音听阈均在正常范围,其中Ⅲ7 1987年(5岁时)曾根据ABR测试结果定为遗传性单耳聋,推测当时可能患有分泌性中耳炎或测试误差等所致。1名72岁配偶4~8 kHz有30~45 dB的双耳对称性感音神经性听力损失,在排除其它可能致聋原因后,考虑为老年聋。
5例初次受检的母系亲属中3例(Ⅲ15、Ⅲ16和Ⅳ1,分别为2、10和6岁)听力正常,另2例(Ⅲ5、Ⅲ6,分别为11岁和8岁)呈双耳对称的高频感音神经性听力损失。
15例复查的母系亲属,除2例各有1耳患中耳炎而表现为不对称性听力损失外,余13例均为双耳对称的感音神经性聋,听力损失发生年龄和程度虽有差异,但高频听力均有下降。耳聋程度:中度2例,重度4例,极重度9例。15名患者中14例有1987年纯音测听结果可进行比较,其中5例9耳听力损失呈进行性加重,另9例11年间听力变化不明显,该2组患者1987年气导听阈显示前者听力损失多数(4例/5例)为轻到中度,而后者多见(8例/9例)于重度和极重度听力损失。6例有AmAn应用史者两次测听结果均为极重度聋,听力损失无明显进展,其中2例仅在低频区检测到残余听力。
, 百拇医药
2.声导抗测试:16名父系亲属及5名配偶声导抗测试结果无异常。
母系亲属中,除2例2耳患中耳炎外,其余受检38耳均为A型鼓室导抗图,其中12耳可在低感觉级(<60 dB SL)引出镫骨肌反射,声反射松弛试验阴性。
3.ABR和TEOAE测试:5例初查的母系亲属中,3例听力正常者均可诱出分化和重复性良好的ABR和TEOAE反应波形;2例有听力损失者ABR反应阈值相应提高,但Ⅰ-Ⅴ波间期或Ⅲ-Ⅴ波间期差在正常范围,TEOAE不能引出,提示为耳蜗性病变。
讨论
线粒体遗传的典型特征是母系遗传,且突变体mtDNA表型的表达具有一定的阈值效应。目前已经公认的与母系遗传性非综合征性耳聋有关的线粒体基因位点有两个:即12S rRNA基因A1555G点突变和tRNA ser(CUN)基因的A7445G点突变[2,4],其中前者与个体对氨基糖甙类抗生素的易感性密切相关[5]。本核心家系1998年调查排除了Ⅲ7为遗传性单耳聋,共发现听力损失者18例,除1例配偶经分析确诊为老年聋外,其余17例患者为母系亲属;利用限制性内切酶Alw26 I消化PCR产物以及mtDNA测序结果表明,家系中所有母系亲属均存在A1555G突变,而男性个体的后代及配偶1 555位碱基正常,也符合母系遗传规律。该结果修正了1987年调查所持常染色体显性遗传聋的观点[1],同时提示mtDNA A1555G突变是该家系致聋的分子基础之一。可能由于线粒体基因突变干扰了内耳正常的氧化磷酸化过程,使三磷酸腺苷ATP生成减少,当积累到一定程度后逐渐导致毛细胞死亡和听力下降。
, 百拇医药
本家系系统听力学测试结果已有报道[1],本次为11年后的抽样追踪。调查显示,在排除中耳炎影响后全部患者表现为双耳对称的感音神经性听力损失;根据部分患者重振试验、ABR和TEOAE测试结果,可将病变部位定位于耳蜗。结合1987年调查及此次复查情况,可以认为该家系患者早期可能仅表现为高频听力损失或以高频下降为主的听力损失,随着年龄增长逐渐向中、低频扩展,呈进行性下降趋势;当听力损失达较重程度时(重度或极重度聋),听力下降趋于缓慢而呈相对固定状态;而应用氨基糖甙类抗生素可使耳聋早发和程度加重。此与其他学者所报道的母系遗传聋听力学特征基本一致[6-8]。
本组结果显示,A1555G突变阳性者并非都存在听力损失,如Ⅲ15、Ⅲ16和Ⅳ1,各患者听力损失发生时间和程度也存在很大差异。据此可以推测,除线粒体基因外可能还有其它因素参与了突变体mtDNA表型表达的调节。AmAn是目前研究最多、最为深入的一种环境调节因子,以往报道的mtDNA A1555G突变致聋多数与此有关[5,8,9]。本家系中6例有常规剂量AmAn应用史的母系亲属全部致聋,而父系亲属及配偶中应用AmAn的9例听力检测均正常,也支持AmAn与A1555G突变致聋存在一定相关性。Prezant等[2]认为人类mtDNA A1555G突变后,其核糖体在结构上更接近大肠杆菌核糖体,从而成为AmAn的理想靶目标。基于这一观点,核心家系中突变个体Ⅲ15、Ⅲ16和Ⅳ1听力正常的原因除与年龄较小、突变体mtDNA尚无足够积累有关外,未接触过氨基糖甙类抗生素可能也是重要因素之一。值得注意的是,本组研究对象全部生活在同一地理环境,且大多数(13例/17例)患者发病前无明确的AmAn应用史,其中2例1987年调查后出生的后代Ⅲ5、Ⅲ6在严格控制耳毒性药物接触后,1998年调查发现也已出现了高频听力损失,表明遗传因素在本家系致聋中占有更为重要的地位。对3名正常表型的突变个体定期进行听力监测将有助于阐明这一观点。
, http://www.100md.com
从系谱分析和听力学检测结果看,本家系与Prezant等[2]报道的阿拉伯-以色列家系有类似之处,后者mtDNA A1555G突变阳性个体间听力损失的差异被认为是由于核基因(常染色体隐性遗传基因)的影响[10]。Fischel-Ghodsian[9]也认为母系遗传聋是多基因遗传决定的,听力损失是否出现遵循阈值学说,即环境因素、线粒体基因和核基因共同作用于靶细胞,使之高于表达阈值时就出现听力损失,反之表型正常。因此,在目前不能用线粒体突变和环境因素圆满解释本家系突变阳性个体间表型差异的情况下,进一步寻找与mtDNA A1555G突变连锁的核基因位点显得尤为重要。
基金项目:国家自然科学基金资助项目(39770402)
通信作者:邢光前
参考文献
1,卜行宽, 殷明德, 曹真, 等. 常染色体显性遗传性感音性聋大家系听力学调查. 中华耳鼻咽喉科杂志, 1992, 27(增刊): 40-43.
, 百拇医药
2,Prezant TR, Agapian JV, Bohlman MC, et al. Mitochondrial ribosomal RNA mutation associated with both antibiotic-induced and non-syndromic deafness. Nat Genet, 1993, 4: 289-294.
3,Report of the informal working group on prevention of deafness and hearing impairment programme planning WHO. Geneva:WHO,1991.
4,Reid FM, Vernham GA, Jacobs HT. A novel mitochondrial point mutation in a maternal pedigree with sensorineural deafness. Hum Mutat, 1994, 3: 243-247.
, 百拇医药
5,袁慧军, 姜泗长, 杨伟炎, 等. 氨基糖甙类抗生素致聋家系线粒体DNA 1555G点突变分析. 中华耳鼻咽喉科杂志, 1998, 33: 67-70.
6,Elverland HH, Torbergsen T. Audiologic findings in a family with mitochondrial disorder. Am J Otol, 1991, 12: 459-465.
7,Usami S, Abe S, Kasai M, et al. Genetic and clinical features of sensorineural hearing loss associated with the 1555 mitochondrial mutation. Laryngoscope, 1997, 107: 483-490.
8,Estivill X, Govea N, Barcelo A, et al. Familial progressive sensorineural deafness is mainly due to the mtDNA A1555G mutation and is enhanced by treatment of aminoglycosides. Am J Hum Genet, 1998, 62: 27-35.
, 百拇医药
9,Fischel-Ghodsian N. Mitochondrial mutations and hearing loss: paradigm for mitochondrial genetics. Am J Hum Genet, 1998, 62: 15-19.
10,Guan MX, Fischel-Ghodsian N, Attardi G. Biochemical evidence for nuclear gene involvement in phenotype of non-syndromic deafness associated with mitochondrial 12S rRNA mutation. Hum Mol Genet, 1996, 5: 963-971.
(收稿日期:1999-05-20), 百拇医药
单位:邢光前(210029 南京医科大学第一附属医院耳鼻咽喉科);卜行宽(210029 南京医科大学第一附属医院耳鼻咽喉科);陆玲(210029 南京医科大学第一附属医院耳鼻咽喉科);杨舜英(210029 南京医科大学第一附属医院耳鼻咽喉科);严明(南京铁道医学院分子生物学研究)
关键词:遗传性疾病;听觉丧失;感音神经性;DNA;线粒体;DNA突变分析;聚合酶链反应
中华耳鼻咽喉科杂志000206 【摘要】 目的 探讨母系遗传性非综合征性耳聋的听力学特征及分子遗传学机制。方法 选择一遗传聋大家系中4代41人为研究对象, 详细询问病史和全身体格检查, 行系统听力学测试和线粒体DNA(mtDNA)A1555G突变分析。结果 20例母系亲属均存在A1555G点突变;纯音测听17例呈程度不等的感音神经性聋,双耳对称,发病年龄1~50岁, 5例听力近11年间呈进行性下降;声导抗测试均为A型鼓室曲线,12耳可在低感觉级引出镫骨肌反射;听性脑干反应( auditory brainstem response,ABR)和瞬态诱发耳声发射测试无蜗后病变证据。21名父系亲属及配偶无听力障碍,mtDNA A1555G突变检测阴性。结论 本家系以迟发性进行性耳蜗性聋为特点,导致听力损失的内在因素为mtDNA A1555G突变,而环境因素和核基因可能也参与了突变体mtDNA表型表达的调节。
, http://www.100md.com
Audiological findings and mitochondrial DNA mutation in a large family with matrilineal sensorineural hearing loss
XING Guangqian, BU Xingkuan, YAN Ming, et al.
(Department of Otorhinolaryngology, First Affiliated Hospital, Nanjing Medical University, Nanjing 210029,China)
Corresponding author: XING Guangqian
【Abstract】 Objective To explore audiological features of matrilineal non-syndromic deafness and its molecular mechanism. Methods A large family with 41 members having inherited deafness was studied. Complete history and the data of general and otolaryngological examinations were collected. All subjects were screened for mitochondrial DNA A1555G mutation by molecular analysis. Audiological evaluation included pure-tone audiometry, auditory brainstem responses and transiently evoked otoacoustic emissions. Results All subjects were in good health generally. Molecular analysis showed that all maternal relatives with or without hearing loss harbored the A1555G mitochondrial mutation. No mutation was found among spouses and paternal relatives. Audiological results showed notable symmetric bilateral sensorineural hearing loss in 17 of 20 maternal relatives, in which 5 cases had a progressive hearing loss in the recent 11 years. The age of appearance of hearing loss ranged from 1 to 50 years. Conclusion All hearing-impaired subjects of this family had late-onset sensorineural hearing loss. Most of which were progressive. The A1555G mitochondrial mutation in the 12S rRNA gene is responsible for the disorder. Other factors, such as nuclear genes or environmental determinants, may influence the clinical expression of mutant mtDNA.
, 百拇医药
【Key words】 Hereditary diseases; Hearing loss,sensorineural; DNA, mitochondrial; DNA mutational analysis; Polymerase chain reaction
1987年我们在江苏省淮阴县发现一遗传聋大家系(6代507人),对其中4代386人进行了遗传学和听力学调查,确诊遗传聋患者137人[1]。为进一步探讨该家系耳聋发生的分子遗传学机制,于1998年3月对其中一分支家系的4代41人进行了系谱复核和系统听力学测试,系谱分析表明该家系耳聋患者呈典型的母系遗传方式传递。同时应用聚合酶链反应(polymerase chain reaction,PCR)产物酶切及PCR产物克隆序列测定技术,对目前文献报道的与非综合征性母系遗传聋相关的mtDNA突变位点――12S rRNA基因的A1555G[2]进行了研究,发现该家系中母系亲属均存在A1555G点突变,而家系配偶及男性个体的后代该突变检测阴性,提示mtDNA A1555G突变是该家系致聋的分子基础之一。
, http://www.100md.com
材料与方法
一、一般资料
该分支家系包括4代41人,其中直系亲属36人(母系亲属20人,父系亲属16人),配偶5人,遗传系谱见图1。母系亲属中原有15人于1987年曾接受过调查,调查内容包括纯音听阈测试和阈上功能检查、声导抗测试及听性脑干反应( auditory brainstem response,ABR)检测;另5人为近年来繁衍后代,属初查。对全体成员行详细病史询问和全身体格检查,尤其注意有无听力下降、眩晕、耳鸣及有无耳毒性药物和噪声接触史,如有听力损失,则详细记录发生年龄及进展情况。
图1 耳聋核心家系图
二、mtDNA A1555G突变分析
, 百拇医药 1.DNA提取及模板mtDNA的扩增:全体成员各抽取外周静脉血5~10 ml,按酚/氯仿抽提法提取DNA。以mtDNA寡核苷酸(nt)1326~1335和(nt)1684~1704为引物,家系成员外周血白细胞粗提DNA为模板,扩增mtDNA (nt)1 326~1 704基因片段。PCR反应参数为:94℃ 4 min变性;94℃ 45 s,60℃ 45 s,72℃ 45 s,共35个循环。2%琼脂糖凝胶电泳观察PCR反应结果。
2.PCR产物纯化和酶切:使用宝灵曼公司PCR产物纯化试剂盒,按使用说明操作,纯化PCR产物。取经纯化的1555位点PCR产物10 μL,加入限制性内切酶Alw26 I (MBI公司,德国)10 U,37℃酶切1 h,2%琼脂糖凝胶电泳观察酶切结果。
3.PCR产物克隆和序列测定:取PCR纯化产物1 μL,T4 DNA连接酶缓冲液1 μL,PEGM-T Vector 1 μL, DNA连接酶1 μL, 双蒸水(ddH2O)6 μL,4℃连接24 h。常规法转化细菌DH5α,涂在含X-gal的LB固体培养基上,37℃培养。选取板上的白色菌落,重新划线培养。单菌落PCR检测,证实有插入片段的阳性克隆后,培养细菌,小制备碱法提取质粒。使用T7启动子引物,377全自动测序仪测序。
, http://www.100md.com
三、听力学检测
除1例2岁的母系亲属听阈由ABR测试结果确定外,全体成员行纯音听阈测试和声导抗测试。纯音听阈以0.5、1、2和4 kHz气导平均值计算,听力损失程度则根据WHO推荐草案[3]确定,即:轻度26~40 dB HL,中度41~60 dB HL,重度61~80 dB HL,极重度≥81 dB HL。进行性听力下降定义为:1987年和本次测试0.5~4 kHz纯音听阈平均提高≥10 dB或其间任意2个频率提高≥15 dB。
对5例初查的母系亲属同时行ABR和瞬态诱发性耳声发射(transiently evoked otoacoustic emission,TEOAE)检查。ABR测试用RION ER-02型诱发电位仪,Ⅴ波反应阈≤30 dB nHL定为正常标准。TEOAE测试用Celesta 503声发射仪进行,刺激声为短声,强度为80 dB peSPL,脉宽80 μs,采用非线性(3+1)给声方式,叠加1 000次,扫描20 ms,延迟5 ms记录,以波形总相关系数r≥0.6为正常标准。
, http://www.100md.com
所有检查在符合要求的隔声室或隔声电磁屏蔽室中进行,仪器均经校准合格使用。
结果
一、一般资料
母系亲属中15例诉听力下降, 均为1987年接受过调查者,根据病史及听力检测结果综合判断其发病年龄为1~50岁。其中10例主诉听力损失进行性加重,5例近11年来继续恶化;4例伴高音调耳鸣;2例有一过性眩晕发作;6例曾应用常规剂量氨基糖甙类抗生素(aminoglycoside antibiotics,AmAn),其中4例(Ⅰ6、Ⅱ11、Ⅲ1、Ⅲ2)在耳聋前3个月内用药,发病年龄1~3岁,另2例应用后原有听力损失急剧加重;2例2耳伴慢性化脓性中耳炎。父系亲属及配偶共21人,9人曾因各种原因接受过AmAn注射治疗。耳鼻咽喉及全身检查无异常。
二、mtDNA 1555位点突变检测结果
, 百拇医药
使用引物mtDNA (nt)1326-1335和(nt)1684-1704扩增家系中所有成员,均可见一379 bp扩增带。由于正常个体mtDNA 1555位点包含有限制性内切酶Alw26 I的切割识别位点GAGA1555C,酶切后可见153 bp和226 bp两条带,当存在A1555G突变时该酶切位点消失,酶切后仅见379 bp一条带。本家系成员酶切结果表明,20例母系亲属均存在mtDNA A1555G突变,而家系中配偶和男性个体(包括男性患者)的后代A1555G突变阴性(图2)。
图2 部分家系成员PCR产物酶切电泳图。M为标准物
对家系成员Ⅰ1、Ⅱ2、Ⅲ1、Ⅳ1 mtDNA 1555位点的测序结果表明,1555位点存在A→G点突变(H链,图3),而配偶和男性后代的1555位碱基为正常的A(L链)。
, 百拇医药
图3 mtDNA 1555位点测序结果(H链)
三、听力学检测结果
1.纯音测听:16名父系亲属和4名配偶纯音听阈均在正常范围,其中Ⅲ7 1987年(5岁时)曾根据ABR测试结果定为遗传性单耳聋,推测当时可能患有分泌性中耳炎或测试误差等所致。1名72岁配偶4~8 kHz有30~45 dB的双耳对称性感音神经性听力损失,在排除其它可能致聋原因后,考虑为老年聋。
5例初次受检的母系亲属中3例(Ⅲ15、Ⅲ16和Ⅳ1,分别为2、10和6岁)听力正常,另2例(Ⅲ5、Ⅲ6,分别为11岁和8岁)呈双耳对称的高频感音神经性听力损失。
15例复查的母系亲属,除2例各有1耳患中耳炎而表现为不对称性听力损失外,余13例均为双耳对称的感音神经性聋,听力损失发生年龄和程度虽有差异,但高频听力均有下降。耳聋程度:中度2例,重度4例,极重度9例。15名患者中14例有1987年纯音测听结果可进行比较,其中5例9耳听力损失呈进行性加重,另9例11年间听力变化不明显,该2组患者1987年气导听阈显示前者听力损失多数(4例/5例)为轻到中度,而后者多见(8例/9例)于重度和极重度听力损失。6例有AmAn应用史者两次测听结果均为极重度聋,听力损失无明显进展,其中2例仅在低频区检测到残余听力。
, 百拇医药
2.声导抗测试:16名父系亲属及5名配偶声导抗测试结果无异常。
母系亲属中,除2例2耳患中耳炎外,其余受检38耳均为A型鼓室导抗图,其中12耳可在低感觉级(<60 dB SL)引出镫骨肌反射,声反射松弛试验阴性。
3.ABR和TEOAE测试:5例初查的母系亲属中,3例听力正常者均可诱出分化和重复性良好的ABR和TEOAE反应波形;2例有听力损失者ABR反应阈值相应提高,但Ⅰ-Ⅴ波间期或Ⅲ-Ⅴ波间期差在正常范围,TEOAE不能引出,提示为耳蜗性病变。
讨论
线粒体遗传的典型特征是母系遗传,且突变体mtDNA表型的表达具有一定的阈值效应。目前已经公认的与母系遗传性非综合征性耳聋有关的线粒体基因位点有两个:即12S rRNA基因A1555G点突变和tRNA ser(CUN)基因的A7445G点突变[2,4],其中前者与个体对氨基糖甙类抗生素的易感性密切相关[5]。本核心家系1998年调查排除了Ⅲ7为遗传性单耳聋,共发现听力损失者18例,除1例配偶经分析确诊为老年聋外,其余17例患者为母系亲属;利用限制性内切酶Alw26 I消化PCR产物以及mtDNA测序结果表明,家系中所有母系亲属均存在A1555G突变,而男性个体的后代及配偶1 555位碱基正常,也符合母系遗传规律。该结果修正了1987年调查所持常染色体显性遗传聋的观点[1],同时提示mtDNA A1555G突变是该家系致聋的分子基础之一。可能由于线粒体基因突变干扰了内耳正常的氧化磷酸化过程,使三磷酸腺苷ATP生成减少,当积累到一定程度后逐渐导致毛细胞死亡和听力下降。
, 百拇医药
本家系系统听力学测试结果已有报道[1],本次为11年后的抽样追踪。调查显示,在排除中耳炎影响后全部患者表现为双耳对称的感音神经性听力损失;根据部分患者重振试验、ABR和TEOAE测试结果,可将病变部位定位于耳蜗。结合1987年调查及此次复查情况,可以认为该家系患者早期可能仅表现为高频听力损失或以高频下降为主的听力损失,随着年龄增长逐渐向中、低频扩展,呈进行性下降趋势;当听力损失达较重程度时(重度或极重度聋),听力下降趋于缓慢而呈相对固定状态;而应用氨基糖甙类抗生素可使耳聋早发和程度加重。此与其他学者所报道的母系遗传聋听力学特征基本一致[6-8]。
本组结果显示,A1555G突变阳性者并非都存在听力损失,如Ⅲ15、Ⅲ16和Ⅳ1,各患者听力损失发生时间和程度也存在很大差异。据此可以推测,除线粒体基因外可能还有其它因素参与了突变体mtDNA表型表达的调节。AmAn是目前研究最多、最为深入的一种环境调节因子,以往报道的mtDNA A1555G突变致聋多数与此有关[5,8,9]。本家系中6例有常规剂量AmAn应用史的母系亲属全部致聋,而父系亲属及配偶中应用AmAn的9例听力检测均正常,也支持AmAn与A1555G突变致聋存在一定相关性。Prezant等[2]认为人类mtDNA A1555G突变后,其核糖体在结构上更接近大肠杆菌核糖体,从而成为AmAn的理想靶目标。基于这一观点,核心家系中突变个体Ⅲ15、Ⅲ16和Ⅳ1听力正常的原因除与年龄较小、突变体mtDNA尚无足够积累有关外,未接触过氨基糖甙类抗生素可能也是重要因素之一。值得注意的是,本组研究对象全部生活在同一地理环境,且大多数(13例/17例)患者发病前无明确的AmAn应用史,其中2例1987年调查后出生的后代Ⅲ5、Ⅲ6在严格控制耳毒性药物接触后,1998年调查发现也已出现了高频听力损失,表明遗传因素在本家系致聋中占有更为重要的地位。对3名正常表型的突变个体定期进行听力监测将有助于阐明这一观点。
, http://www.100md.com
从系谱分析和听力学检测结果看,本家系与Prezant等[2]报道的阿拉伯-以色列家系有类似之处,后者mtDNA A1555G突变阳性个体间听力损失的差异被认为是由于核基因(常染色体隐性遗传基因)的影响[10]。Fischel-Ghodsian[9]也认为母系遗传聋是多基因遗传决定的,听力损失是否出现遵循阈值学说,即环境因素、线粒体基因和核基因共同作用于靶细胞,使之高于表达阈值时就出现听力损失,反之表型正常。因此,在目前不能用线粒体突变和环境因素圆满解释本家系突变阳性个体间表型差异的情况下,进一步寻找与mtDNA A1555G突变连锁的核基因位点显得尤为重要。
基金项目:国家自然科学基金资助项目(39770402)
通信作者:邢光前
参考文献
1,卜行宽, 殷明德, 曹真, 等. 常染色体显性遗传性感音性聋大家系听力学调查. 中华耳鼻咽喉科杂志, 1992, 27(增刊): 40-43.
, 百拇医药
2,Prezant TR, Agapian JV, Bohlman MC, et al. Mitochondrial ribosomal RNA mutation associated with both antibiotic-induced and non-syndromic deafness. Nat Genet, 1993, 4: 289-294.
3,Report of the informal working group on prevention of deafness and hearing impairment programme planning WHO. Geneva:WHO,1991.
4,Reid FM, Vernham GA, Jacobs HT. A novel mitochondrial point mutation in a maternal pedigree with sensorineural deafness. Hum Mutat, 1994, 3: 243-247.
, 百拇医药
5,袁慧军, 姜泗长, 杨伟炎, 等. 氨基糖甙类抗生素致聋家系线粒体DNA 1555G点突变分析. 中华耳鼻咽喉科杂志, 1998, 33: 67-70.
6,Elverland HH, Torbergsen T. Audiologic findings in a family with mitochondrial disorder. Am J Otol, 1991, 12: 459-465.
7,Usami S, Abe S, Kasai M, et al. Genetic and clinical features of sensorineural hearing loss associated with the 1555 mitochondrial mutation. Laryngoscope, 1997, 107: 483-490.
8,Estivill X, Govea N, Barcelo A, et al. Familial progressive sensorineural deafness is mainly due to the mtDNA A1555G mutation and is enhanced by treatment of aminoglycosides. Am J Hum Genet, 1998, 62: 27-35.
, 百拇医药
9,Fischel-Ghodsian N. Mitochondrial mutations and hearing loss: paradigm for mitochondrial genetics. Am J Hum Genet, 1998, 62: 15-19.
10,Guan MX, Fischel-Ghodsian N, Attardi G. Biochemical evidence for nuclear gene involvement in phenotype of non-syndromic deafness associated with mitochondrial 12S rRNA mutation. Hum Mol Genet, 1996, 5: 963-971.
(收稿日期:1999-05-20), 百拇医药