脂质沉积性肌病15例临床及病理分析
作者:林红 王柠 慕容慎行 张文敏 陈振斌
单位:林红(福建医科大学附属第一医院神经内科,福州 350004); 王柠(福建医科大学附属第一医院神经内科,福州 350004); 慕容慎行(福建医科大学附属第一医院神经内科,福州 350004); 张文敏(福建医科大学病理解剖学教研室,福州 350004); 陈振斌(福建医科大学电子显微镜室,福州 350004)
关键词:脂质累积病;肌疾病,病理学;肉碱
福建医科大学学报000117 摘要:目的 结合肌肉病理改变,探讨脂质沉积性肌病(LSM)的诊断及治疗。方法 对15例LSM患者进行临床及病理分析,光镜与电镜观察。结果 患者均有进行性四肢无力,血清肌酶不同程度升高,肌肉病理苏丹黑染色及电镜示肌纤维中大量脂质沉积,以Ⅰ型纤维为主。结论 LSM是一组以肌无力和肌疲劳为主要表现的肌纤维内脂肪代谢障碍病。肌肉病理检查有助于确定诊断。激素治疗有效。
, http://www.100md.com
中图分类号:R746 文献标识码:A
文章编号:1000-2235(2000)01-0050-03
Clinical and Pathological Analysis of Lipid Storage Myopathy(Report of 15 Cases)
LIN Hong,WANG Ning,MURONG Shen-xing,et al
(Department of Neurology,The Affiliated Hospital,Fujian Medical University,Fuzhou 350005,China)
ABSTRACT:Objective To study 15 cases of lipid storage myopathy(LSM) and to analyze them in clinical and pathological features.Methods The clinical data of 15 cases were collected and muscle biopsies were done with routine and histochemical staining.Light and electron microscopic studies were made.Results The clinical characters of all patients were progressive muscle weakness with increased muscle enzymes in the plasma.The light and electron microscopic studies of muscle biopsy materials showed excessive amounts of fatty droplets in muscle fibers.Type I muscle fibers are more severely affected by the storage of the fat.Conclusion Fatigue and muscle weakness especially in proximal muscle were prominent symptoms in LSM.Muscle biopsy has a significant value for the definite diagnosis of this disease.LSM can be treated by corticosteroid.
, 百拇医药
KEY WORDS:lipidosis;muscular diseases,pathlogy;carnitine
脂质沉积性肌病(LSM)是由于肌肉中长链脂肪酸代谢障碍导致脂质沉积在肌纤维中而引起的肌病。临床主要表现为四肢无力,近端为主,且活动后易疲劳,症状可进行性加重,也可时轻时重,有波动性,或有缓解复发倾向。本病临床常见,但常被误诊为多发性肌炎、肢带型肌营养不良症及重症肌无力等。其肌肉病理和电镜观察见肌肉纤维中大量脂质沉积的特征性改变,有助于诊断[1]。我科自1994~1999年收治15例LSM,均经肌肉活检病理证实,现将其临床特点、肌肉组化和电镜改变以及治疗总结报告如下,以提高对本病的认识。
1 临床资料
1.1 一般资料 全组15例,男性5例,女性10例,男∶女比为1∶2,年龄21.6±4.87岁(12~40岁)。其中20岁以下11例,20~40岁4例。病程6~48个月。
, http://www.100md.com
15例均为缓慢起病,主要为骨骼肌受累,表现为四肢无力,以近端肢体为著,活动后易疲劳,伴肌肉酸痛。4例肌无力呈进行性加重;8例症状时轻时重,有波动性;3例有明显的缓解复发。12例伴有颈肌无力;8例伴咀嚼肌无力;6例伴吞咽肌无力;4例伴四肢近端轻度肌肉萎缩;1例四肢末梢套型感觉障碍;1例伴有心悸感;2例肝脏弥漫性改变伴肝肿大;2例可疑家族史。
1.2 实验室检查 15例病人血清肌酸磷酸激酶(CK)均增高,其中250~800 U/L 8例,1250~6975 U/L 6例,正常1例(正常值25~200 U/L);乳酸脱氢酶(LDH)250~600 U/L 8例,1000~2100 U/L 7例(正常值109~245 U/L)。13例血清肌红蛋白(Mb)>250 μg/L(正常值110 μg/L),2例未检;15例谷-草转氨酶(GOT)70~219 U/L(正常值8~40 U/L);6例高密度脂蛋白胆固醇(HDLC)增高1.79~2.49 U/L(正常值1.1~1.7 U/L);1例总胆固醇6.92 mmol/L(正常值3.6~6.4 mmol/L)。肌电图检查肌源性损害14例,1例呈神经源性损害。15例中11例查心电图正常或大致正常。周围血白细胞涂片苏丹黑染色7例,3例可见白细胞内脂滴明显增多。
, 百拇医药
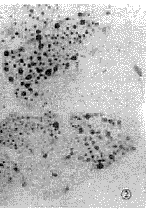
1.3 肌肉病理及电镜检查 15例均行肱二头肌或股四头肌肌肉活检。标本行苏木素-伊红染色、苏丹黑染色以及ATP酶染色、还原酶辅酶Ⅰ染色。15例患者苏木素-伊红染色骨骼肌肌纤维内均有大量脂滴沉积,呈圆形空泡,部分相互融合(图1)。苏丹黑染色显示肌纤维内充满黑色空泡,为脂肪滴(图2)。ATP酶染色显示脂滴沉积以Ⅰ型肌纤维为主。电镜检查: 15例病人于肌原纤维间或质膜下见脂滴明显增多,成串或成簇堆积(图3),多位于线粒体附近,肌丝及肌节结构尚规则,其中4例伴线粒体数量增多。未见类晶体颗粒及破碎红纤维。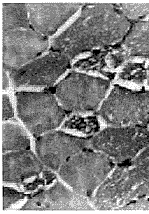

图1 肌纤维内脂滴呈空泡样,部分相互融合.HE ×200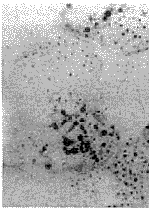
, http://www.100md.com
图2 肌纤维内大量脂肪滴聚集,呈空泡样改变.苏丹黑染色 ×200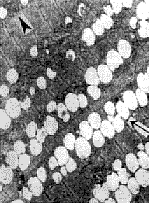

图3 肌细胞内增多的脂滴呈串珠状排列于肌原纤维间或质膜下,线粒体增多.×4500
1.4 诊断与治疗 本组病例初期多数被误诊,其中6例误诊为多发性肌炎,2例误诊为肢带型肌营养不良症,1例误诊为慢性格林-巴利综合征。经肌肉活检,组化染色及电镜检查后明确诊断。治疗上采用强的松20~50 mg/d,每日晨一次顿服或甲基强的松龙40~60 mg/d,静脉滴注,症状好转后改口服,激素缓慢减量,至5~10 mg/d维持6个月,同时辅以ATP、辅酶A及肌苷片等药物,配合低脂、高碳水化合物饮食。15例患者临床症状及血清肌酶谱均有明显改善或基本恢复。其中9例于治疗后2周症状开始好转,6例于4周后开始好转。3例病人于症状好转后自行停药,分别于4个月、2年、4年后症状再发,经激素治疗后恢复。
, 百拇医药
2 讨 论
2.1 诊断及分型 LSM是一种由于生化缺陷直接或间接地引起肌纤维脂肪酸代谢障碍病[2]。根据生化缺陷,LSM可分为肌肉肉毒碱缺乏症、全身肉毒碱缺乏症和肉毒碱软脂酰转移酶缺乏症[3]。
(1)肌肉肉毒碱缺乏症: 多于儿童或青少年发病,其肌肉肉毒碱含量显著降低,而血浆中肉毒碱浓度正常。表现为四肢近端无力,活动后易乏力。肌肉活检显示肌浆中过量脂滴空泡聚积,以Ⅰ型肌纤维受累为著。血清肌酶升高。激素治疗有效。(2)全身肉毒碱缺乏症: 为常染色体隐性遗传病,血浆、肌肉、肝脏中肉毒碱浓度均降低,除表现为肢体无力外,多有反复发作性肝脏脑病,或有低血糖或心肌病。肉毒碱治疗有效。全身肉毒碱缺乏症亦可为继发性,多继发于营养不良、肝脏疾病及长期血透病人中。(3)肉毒碱软脂酰转移酶缺乏症: 表现为痛性肌痉挛和发作性肌红蛋白尿。病理于发作期见肌纤维大片坏死,Ⅰ型纤维明显空泡形成[4]。肌肉生化检测肉毒碱软脂酰转移酶降低。肉毒碱治疗有效。(4)近年来还发现乙酰辅酶A脱氢酶缺乏也可致LSM,低脂饮食可减少发作。Papadimitriou报道1例乙酰辅酶A脱氢酶明显缺乏致LSM,病因考虑可能为服用丙戊酸钠所致,核黄素治疗有效[5]。
, http://www.100md.com
本组15例临床表现仅局限于骨骼肌无力,无肝性脑病、低血糖、心肌病及肌红蛋白尿等全身性症状。肌肉病理及电镜观察肌纤维中脂质沉积,以Ⅰ型纤维受累为主,电镜下,脂滴聚积在线粒体周围,部分病例尚有线粒体增多,诊断LSM明确。患者虽未进行肌肉生化检测,但结合临床及激素治疗有效,估计由肌肉肉毒碱缺乏所致的LSM可能性最大。本组查7例患者血白细胞苏丹黑染色,3例见白细胞内大量脂滴聚集,其中2例白细胞内脂滴随病情好转而减少,考虑白细胞内脂质沉积与病情轻重有关。此项检查对LSM预后观察是否有指导意义需进一步探讨。
LSM临床上易误诊为多发性肌炎、肢带型肌营养不良症或慢性格林-巴利综合征,肌肉病理可以鉴别。作者认为凡四肢近端无力患者,慢性或亚急性起病,尤其活动后易疲劳、症状时轻时重者,均需行肌肉活检,予组化染色及电镜检查,以免误诊。本组病例中有1例出现四肢套型感觉障碍,肌电图呈神经源性损害,LSM出现周围神经损害,考虑与脂质累及雪旺细胞有关[6]。
, http://www.100md.com 2.2 治疗 LSM治疗以强的松或强的松龙为主,笔者采用强的松20~60 mg每日晨一次顿服,平均40 mg,同时辅以ATP、辅酶A及肌苷等药物治疗,予高碳水化合物及低脂饮食,多进食羊肉、牛肉等富含肉毒碱食物。经治疗,15例LSM症状及血清肌酶均有明显改善或基本恢复,说明激素使用有效。
激素治疗LSM的机制尚不明确,可能与激素有利于长链脂肪酸进入线粒体及其氧化增强或对三酸甘油酯的直接激活作用有关[6]。本组3例激素治疗后症状恢复,停药后复发,考虑与激素减量及停药过快有关。建议使用强的松20~40 mg/d,症状好转后应缓慢减量,至5~10 mg/d,应维持半年以上,以减少复发。文献报道强的松治疗疗效不佳者,可合用或单独服用普萘洛尔40 mg 3/d,其机制尚不明确[7]。
作者简介:林 红,女,1965年2月生,主治医师.
参考文献:
, 百拇医药
[1]陈 琳,郭 重,郭玉璞,等.脂质沉积性肌病的临床和病理特点[J].中华神经科杂志,1998;31(3):165~167
[2]Munoz-Blanco JL.Lipidic myopathies[J].Rev Neurol,1998;(Suppl):72~80
[3]Di Mauro S,Trevisan C,Hays A.Disorders of lipid metabolism in muscle[J].Muscle Nerve,1980;3(5):369~388
[4]陈碧芬.骨骼肌疾病病理学[M].福州:福建科学技术出版社,1993: 96~99
[5]Papadimitriou A,Servidei S.Late onset lipid storage myopathy due to multiple acyl CoA dehydrogenase deficiency triggered by valproate[J].Neuromuscul Disord,1991;1(4):247~252
[6]Engel AG,Robert GS,Rochester S.Lipid storage myopathy response to prednisone[J].Arch Neurol,1972;27:174~181
[7]Martyn C,Jelliuck EH,Webb JN.Lipid storage myopathy:successful treatment with propronolol[J].Br Med J,1981;20:1997~1999
收稿日期:2000-01-26, http://www.100md.com
单位:林红(福建医科大学附属第一医院神经内科,福州 350004); 王柠(福建医科大学附属第一医院神经内科,福州 350004); 慕容慎行(福建医科大学附属第一医院神经内科,福州 350004); 张文敏(福建医科大学病理解剖学教研室,福州 350004); 陈振斌(福建医科大学电子显微镜室,福州 350004)
关键词:脂质累积病;肌疾病,病理学;肉碱
福建医科大学学报000117 摘要:目的 结合肌肉病理改变,探讨脂质沉积性肌病(LSM)的诊断及治疗。方法 对15例LSM患者进行临床及病理分析,光镜与电镜观察。结果 患者均有进行性四肢无力,血清肌酶不同程度升高,肌肉病理苏丹黑染色及电镜示肌纤维中大量脂质沉积,以Ⅰ型纤维为主。结论 LSM是一组以肌无力和肌疲劳为主要表现的肌纤维内脂肪代谢障碍病。肌肉病理检查有助于确定诊断。激素治疗有效。
, http://www.100md.com
中图分类号:R746 文献标识码:A
文章编号:1000-2235(2000)01-0050-03
Clinical and Pathological Analysis of Lipid Storage Myopathy(Report of 15 Cases)
LIN Hong,WANG Ning,MURONG Shen-xing,et al
(Department of Neurology,The Affiliated Hospital,Fujian Medical University,Fuzhou 350005,China)
ABSTRACT:Objective To study 15 cases of lipid storage myopathy(LSM) and to analyze them in clinical and pathological features.Methods The clinical data of 15 cases were collected and muscle biopsies were done with routine and histochemical staining.Light and electron microscopic studies were made.Results The clinical characters of all patients were progressive muscle weakness with increased muscle enzymes in the plasma.The light and electron microscopic studies of muscle biopsy materials showed excessive amounts of fatty droplets in muscle fibers.Type I muscle fibers are more severely affected by the storage of the fat.Conclusion Fatigue and muscle weakness especially in proximal muscle were prominent symptoms in LSM.Muscle biopsy has a significant value for the definite diagnosis of this disease.LSM can be treated by corticosteroid.
, 百拇医药
KEY WORDS:lipidosis;muscular diseases,pathlogy;carnitine
脂质沉积性肌病(LSM)是由于肌肉中长链脂肪酸代谢障碍导致脂质沉积在肌纤维中而引起的肌病。临床主要表现为四肢无力,近端为主,且活动后易疲劳,症状可进行性加重,也可时轻时重,有波动性,或有缓解复发倾向。本病临床常见,但常被误诊为多发性肌炎、肢带型肌营养不良症及重症肌无力等。其肌肉病理和电镜观察见肌肉纤维中大量脂质沉积的特征性改变,有助于诊断[1]。我科自1994~1999年收治15例LSM,均经肌肉活检病理证实,现将其临床特点、肌肉组化和电镜改变以及治疗总结报告如下,以提高对本病的认识。
1 临床资料
1.1 一般资料 全组15例,男性5例,女性10例,男∶女比为1∶2,年龄21.6±4.87岁(12~40岁)。其中20岁以下11例,20~40岁4例。病程6~48个月。
, http://www.100md.com
15例均为缓慢起病,主要为骨骼肌受累,表现为四肢无力,以近端肢体为著,活动后易疲劳,伴肌肉酸痛。4例肌无力呈进行性加重;8例症状时轻时重,有波动性;3例有明显的缓解复发。12例伴有颈肌无力;8例伴咀嚼肌无力;6例伴吞咽肌无力;4例伴四肢近端轻度肌肉萎缩;1例四肢末梢套型感觉障碍;1例伴有心悸感;2例肝脏弥漫性改变伴肝肿大;2例可疑家族史。
1.2 实验室检查 15例病人血清肌酸磷酸激酶(CK)均增高,其中250~800 U/L 8例,1250~6975 U/L 6例,正常1例(正常值25~200 U/L);乳酸脱氢酶(LDH)250~600 U/L 8例,1000~2100 U/L 7例(正常值109~245 U/L)。13例血清肌红蛋白(Mb)>250 μg/L(正常值110 μg/L),2例未检;15例谷-草转氨酶(GOT)70~219 U/L(正常值8~40 U/L);6例高密度脂蛋白胆固醇(HDLC)增高1.79~2.49 U/L(正常值1.1~1.7 U/L);1例总胆固醇6.92 mmol/L(正常值3.6~6.4 mmol/L)。肌电图检查肌源性损害14例,1例呈神经源性损害。15例中11例查心电图正常或大致正常。周围血白细胞涂片苏丹黑染色7例,3例可见白细胞内脂滴明显增多。
, 百拇医药
1.3 肌肉病理及电镜检查 15例均行肱二头肌或股四头肌肌肉活检。标本行苏木素-伊红染色、苏丹黑染色以及ATP酶染色、还原酶辅酶Ⅰ染色。15例患者苏木素-伊红染色骨骼肌肌纤维内均有大量脂滴沉积,呈圆形空泡,部分相互融合(图1)。苏丹黑染色显示肌纤维内充满黑色空泡,为脂肪滴(图2)。ATP酶染色显示脂滴沉积以Ⅰ型肌纤维为主。电镜检查: 15例病人于肌原纤维间或质膜下见脂滴明显增多,成串或成簇堆积(图3),多位于线粒体附近,肌丝及肌节结构尚规则,其中4例伴线粒体数量增多。未见类晶体颗粒及破碎红纤维。
图1 肌纤维内脂滴呈空泡样,部分相互融合.HE ×200
, http://www.100md.com
图2 肌纤维内大量脂肪滴聚集,呈空泡样改变.苏丹黑染色 ×200
图3 肌细胞内增多的脂滴呈串珠状排列于肌原纤维间或质膜下,线粒体增多.×4500
1.4 诊断与治疗 本组病例初期多数被误诊,其中6例误诊为多发性肌炎,2例误诊为肢带型肌营养不良症,1例误诊为慢性格林-巴利综合征。经肌肉活检,组化染色及电镜检查后明确诊断。治疗上采用强的松20~50 mg/d,每日晨一次顿服或甲基强的松龙40~60 mg/d,静脉滴注,症状好转后改口服,激素缓慢减量,至5~10 mg/d维持6个月,同时辅以ATP、辅酶A及肌苷片等药物,配合低脂、高碳水化合物饮食。15例患者临床症状及血清肌酶谱均有明显改善或基本恢复。其中9例于治疗后2周症状开始好转,6例于4周后开始好转。3例病人于症状好转后自行停药,分别于4个月、2年、4年后症状再发,经激素治疗后恢复。
, 百拇医药
2 讨 论
2.1 诊断及分型 LSM是一种由于生化缺陷直接或间接地引起肌纤维脂肪酸代谢障碍病[2]。根据生化缺陷,LSM可分为肌肉肉毒碱缺乏症、全身肉毒碱缺乏症和肉毒碱软脂酰转移酶缺乏症[3]。
(1)肌肉肉毒碱缺乏症: 多于儿童或青少年发病,其肌肉肉毒碱含量显著降低,而血浆中肉毒碱浓度正常。表现为四肢近端无力,活动后易乏力。肌肉活检显示肌浆中过量脂滴空泡聚积,以Ⅰ型肌纤维受累为著。血清肌酶升高。激素治疗有效。(2)全身肉毒碱缺乏症: 为常染色体隐性遗传病,血浆、肌肉、肝脏中肉毒碱浓度均降低,除表现为肢体无力外,多有反复发作性肝脏脑病,或有低血糖或心肌病。肉毒碱治疗有效。全身肉毒碱缺乏症亦可为继发性,多继发于营养不良、肝脏疾病及长期血透病人中。(3)肉毒碱软脂酰转移酶缺乏症: 表现为痛性肌痉挛和发作性肌红蛋白尿。病理于发作期见肌纤维大片坏死,Ⅰ型纤维明显空泡形成[4]。肌肉生化检测肉毒碱软脂酰转移酶降低。肉毒碱治疗有效。(4)近年来还发现乙酰辅酶A脱氢酶缺乏也可致LSM,低脂饮食可减少发作。Papadimitriou报道1例乙酰辅酶A脱氢酶明显缺乏致LSM,病因考虑可能为服用丙戊酸钠所致,核黄素治疗有效[5]。
, http://www.100md.com
本组15例临床表现仅局限于骨骼肌无力,无肝性脑病、低血糖、心肌病及肌红蛋白尿等全身性症状。肌肉病理及电镜观察肌纤维中脂质沉积,以Ⅰ型纤维受累为主,电镜下,脂滴聚积在线粒体周围,部分病例尚有线粒体增多,诊断LSM明确。患者虽未进行肌肉生化检测,但结合临床及激素治疗有效,估计由肌肉肉毒碱缺乏所致的LSM可能性最大。本组查7例患者血白细胞苏丹黑染色,3例见白细胞内大量脂滴聚集,其中2例白细胞内脂滴随病情好转而减少,考虑白细胞内脂质沉积与病情轻重有关。此项检查对LSM预后观察是否有指导意义需进一步探讨。
LSM临床上易误诊为多发性肌炎、肢带型肌营养不良症或慢性格林-巴利综合征,肌肉病理可以鉴别。作者认为凡四肢近端无力患者,慢性或亚急性起病,尤其活动后易疲劳、症状时轻时重者,均需行肌肉活检,予组化染色及电镜检查,以免误诊。本组病例中有1例出现四肢套型感觉障碍,肌电图呈神经源性损害,LSM出现周围神经损害,考虑与脂质累及雪旺细胞有关[6]。
, http://www.100md.com 2.2 治疗 LSM治疗以强的松或强的松龙为主,笔者采用强的松20~60 mg每日晨一次顿服,平均40 mg,同时辅以ATP、辅酶A及肌苷等药物治疗,予高碳水化合物及低脂饮食,多进食羊肉、牛肉等富含肉毒碱食物。经治疗,15例LSM症状及血清肌酶均有明显改善或基本恢复,说明激素使用有效。
激素治疗LSM的机制尚不明确,可能与激素有利于长链脂肪酸进入线粒体及其氧化增强或对三酸甘油酯的直接激活作用有关[6]。本组3例激素治疗后症状恢复,停药后复发,考虑与激素减量及停药过快有关。建议使用强的松20~40 mg/d,症状好转后应缓慢减量,至5~10 mg/d,应维持半年以上,以减少复发。文献报道强的松治疗疗效不佳者,可合用或单独服用普萘洛尔40 mg 3/d,其机制尚不明确[7]。
作者简介:林 红,女,1965年2月生,主治医师.
参考文献:
, 百拇医药
[1]陈 琳,郭 重,郭玉璞,等.脂质沉积性肌病的临床和病理特点[J].中华神经科杂志,1998;31(3):165~167
[2]Munoz-Blanco JL.Lipidic myopathies[J].Rev Neurol,1998;(Suppl):72~80
[3]Di Mauro S,Trevisan C,Hays A.Disorders of lipid metabolism in muscle[J].Muscle Nerve,1980;3(5):369~388
[4]陈碧芬.骨骼肌疾病病理学[M].福州:福建科学技术出版社,1993: 96~99
[5]Papadimitriou A,Servidei S.Late onset lipid storage myopathy due to multiple acyl CoA dehydrogenase deficiency triggered by valproate[J].Neuromuscul Disord,1991;1(4):247~252
[6]Engel AG,Robert GS,Rochester S.Lipid storage myopathy response to prednisone[J].Arch Neurol,1972;27:174~181
[7]Martyn C,Jelliuck EH,Webb JN.Lipid storage myopathy:successful treatment with propronolol[J].Br Med J,1981;20:1997~1999
收稿日期:2000-01-26, http://www.100md.com