高效液相色谱法测定克拉霉素干混悬剂中克拉霉素的含量
http://www.100md.com
37c医学网
作者:苏茵
单位:广州市药品检验所,广东 广州 510160
关键词:高效液相色谱法;克拉霉素;药物分析
广东药学院学报000311
摘 要 采用RP-HPLC法测定克拉霉素干混悬剂中克拉霉素的含量。色谱柱为μ-Bondapark C18柱,流动相为pH 6.0的V(0.067 mol/L磷酸二氢钾):V(乙腈):V(甲醇)=63∶32∶5,检测波长210 nm。在100~700 μg/mL范围内,浓度与峰面积线性关系良好(r=0.9992)。克拉霉素的平均回收率为101.7%,RSD=1.1%,n=6。方法简便,精密度好。
中图号 R927.2 文献标识码:A
, 百拇医药 文章编号:1006-8783(2000)03-0201-02
克拉霉素干混悬剂是半合成红霉素衍生物克拉霉素的一种新剂型。具药物吸收良好,受食物影响不大,利于婴儿、儿童服用等优点[1]。在美国药典24版、日抗基等均有收载。文献[1]所采用的HPLC法,流动相均为甲醇-pH 3.5缓冲液,由于末端吸收在210 nm处,甲醇吸收大,测定基线不稳,且流动相酸性较大,对色谱柱损坏较大。本文通过改变流动相的配比和pH值,使基线稳定,精密度好,且辅料及杂质降解物不干扰测定,测定结果满意。
1 实验部分
1.1 仪器与试药
日本岛津高效液相色谱仪ClassLC-10A,SPD-10A紫外检测器,C-R6A数据处理机。
克拉霉素对照品、克拉霉素干混悬剂供试品(均由南新药业有限公司提供),乙腈、甲醇(色谱纯),磷酸二氢钾(分析纯)。
, 百拇医药
1.2 色谱条件 色谱柱,μ-Bondapak, C18柱(10 μm,4.6 mm×200 mm,大连化物所);流动相,V(0.067 mol/L磷酸二氢钾):V(乙腈):V(甲醇)=63∶32∶5,调pH至6.0;流速,2.0 mL/min;检测波长,210 nm;进样量,20 μL;柱温,50 ℃。
2 实验结果
2.1 线性范围
精密称取克拉霉素对照品125 mg,置50 mL量瓶中,加10 mL乙腈使溶解,加流动相至刻度,分别精密吸取1.0、2.0、3.0、5.0、7.0 mL,置25 mL量瓶中,用流动相稀释至刻度,制成100、200、300、500、700 μg/mL的溶液,按上述色谱条件进样20 μL,记录峰面积。以质量浓度对峰面积进行线性回归,得回归方程Y=10190.3X+582.6,r=0.9992。表明克拉霉素浓度在100~700 μg/mL范围内与峰面积线性关系良好。
, 百拇医药
2.2 回收率试验
取相同量的克拉霉素干混悬剂样品和对照品,按样品测定项下方法溶解,配成250 μg/mL溶液,测得样品的加样回收率为100.1%、103.2%、102.0%、102.3%、101.2%、100.6%,平均加样回收率为101.7%(n=6),RSD=1.1%。
2.3 精密度、重复性和稳定性试验
取同一供试液按上述色谱条件连续进样5次,峰面积基本不变,RSD为1.0%。取同一批号样品5份,分别按样品测定项下方法进行测定,RSD为0.62%。取同一供试液分别于0、2、4、6、8 h进行测定,峰面积基本不变,RSD为1.1%。
2.4 样品测定
取供试品适量(相当于克拉霉素125 mg),置500 mL量瓶,加乙腈50 mL,超声溶解,加磷酸二氢钾缓冲液34 mL,超声溶解30 min,加甲醇至刻度,滤过。另取克拉霉素对照品适量,加乙腈适量,用流动相配成250 μg/mL的溶液,在上述色谱条件下分别进样20 μL,按外标法对3批样品进行测定,并与微生物法比较,结果见表1,对两种方法的测定结果进行t检验,结果两者差异不显著(P>0.05)。
, 百拇医药
表1 样品含量测定结果(n=2,%) 批号
HPLC法
微生物法
991101
99.97
100.2
991102
100.5
99.3
991103
97.50
98.9
, 百拇医药
3 讨论
3.1 本法色谱图见图1,克拉霉素保留时间为6.5 min,干混悬剂中其他辅料对测定不干扰。取供试品在150 ℃干热条件下进行降解破坏,其降解后色谱图见图2,降解峰与供试品峰的分离度达2.7,可有效分离,不影响供试品测定。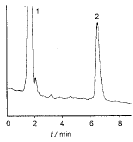
图1 供试品的色谱图
1 辅料 2 克拉霉素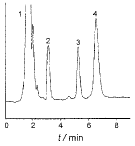
图2 150 ℃干热降解色谱图
1 辅料 2、3 降解峰 4 克拉霉素
3.2 由于克拉霉素不溶于水,其干混悬剂中又含有大量的辅料,故采用乙腈溶解后,加入磷酸盐缓冲液及甲醇,进行超声溶解,以此配制成的溶液,经实验,室温放置8 h,含量仍稳定。
, 百拇医药
3.3 大环内酯类抗生素本身紫外吸收不明显,HPLC法多采用末端吸收波长210 nm,由于甲醇吸收大,若移动相中采用40%的甲醇,基线测定不稳,重现性差。经实验,移动相按V(0.067 mol/L磷酸二氢钾):V(乙腈):V(甲醇)=63∶32∶5的配比,既能得到稳定的基线,又达到有效的分离,结果满意。
参考文献
[1]Gan VN,Chu SY,Kuamiesz HT, et al. Pharmacokinetics of a clarithromycin suspension in Infants and children[J]. AAC,1992,36(11):2478
[2]USP 24[S].425.
(收稿日期 2000-05-22), 百拇医药
单位:广州市药品检验所,广东 广州 510160
关键词:高效液相色谱法;克拉霉素;药物分析
广东药学院学报000311
摘 要 采用RP-HPLC法测定克拉霉素干混悬剂中克拉霉素的含量。色谱柱为μ-Bondapark C18柱,流动相为pH 6.0的V(0.067 mol/L磷酸二氢钾):V(乙腈):V(甲醇)=63∶32∶5,检测波长210 nm。在100~700 μg/mL范围内,浓度与峰面积线性关系良好(r=0.9992)。克拉霉素的平均回收率为101.7%,RSD=1.1%,n=6。方法简便,精密度好。
中图号 R927.2 文献标识码:A
, 百拇医药 文章编号:1006-8783(2000)03-0201-02
克拉霉素干混悬剂是半合成红霉素衍生物克拉霉素的一种新剂型。具药物吸收良好,受食物影响不大,利于婴儿、儿童服用等优点[1]。在美国药典24版、日抗基等均有收载。文献[1]所采用的HPLC法,流动相均为甲醇-pH 3.5缓冲液,由于末端吸收在210 nm处,甲醇吸收大,测定基线不稳,且流动相酸性较大,对色谱柱损坏较大。本文通过改变流动相的配比和pH值,使基线稳定,精密度好,且辅料及杂质降解物不干扰测定,测定结果满意。
1 实验部分
1.1 仪器与试药
日本岛津高效液相色谱仪ClassLC-10A,SPD-10A紫外检测器,C-R6A数据处理机。
克拉霉素对照品、克拉霉素干混悬剂供试品(均由南新药业有限公司提供),乙腈、甲醇(色谱纯),磷酸二氢钾(分析纯)。
, 百拇医药
1.2 色谱条件 色谱柱,μ-Bondapak, C18柱(10 μm,4.6 mm×200 mm,大连化物所);流动相,V(0.067 mol/L磷酸二氢钾):V(乙腈):V(甲醇)=63∶32∶5,调pH至6.0;流速,2.0 mL/min;检测波长,210 nm;进样量,20 μL;柱温,50 ℃。
2 实验结果
2.1 线性范围
精密称取克拉霉素对照品125 mg,置50 mL量瓶中,加10 mL乙腈使溶解,加流动相至刻度,分别精密吸取1.0、2.0、3.0、5.0、7.0 mL,置25 mL量瓶中,用流动相稀释至刻度,制成100、200、300、500、700 μg/mL的溶液,按上述色谱条件进样20 μL,记录峰面积。以质量浓度对峰面积进行线性回归,得回归方程Y=10190.3X+582.6,r=0.9992。表明克拉霉素浓度在100~700 μg/mL范围内与峰面积线性关系良好。
, 百拇医药
2.2 回收率试验
取相同量的克拉霉素干混悬剂样品和对照品,按样品测定项下方法溶解,配成250 μg/mL溶液,测得样品的加样回收率为100.1%、103.2%、102.0%、102.3%、101.2%、100.6%,平均加样回收率为101.7%(n=6),RSD=1.1%。
2.3 精密度、重复性和稳定性试验
取同一供试液按上述色谱条件连续进样5次,峰面积基本不变,RSD为1.0%。取同一批号样品5份,分别按样品测定项下方法进行测定,RSD为0.62%。取同一供试液分别于0、2、4、6、8 h进行测定,峰面积基本不变,RSD为1.1%。
2.4 样品测定
取供试品适量(相当于克拉霉素125 mg),置500 mL量瓶,加乙腈50 mL,超声溶解,加磷酸二氢钾缓冲液34 mL,超声溶解30 min,加甲醇至刻度,滤过。另取克拉霉素对照品适量,加乙腈适量,用流动相配成250 μg/mL的溶液,在上述色谱条件下分别进样20 μL,按外标法对3批样品进行测定,并与微生物法比较,结果见表1,对两种方法的测定结果进行t检验,结果两者差异不显著(P>0.05)。
, 百拇医药
表1 样品含量测定结果(n=2,%) 批号
HPLC法
微生物法
991101
99.97
100.2
991102
100.5
99.3
991103
97.50
98.9
, 百拇医药
3 讨论
3.1 本法色谱图见图1,克拉霉素保留时间为6.5 min,干混悬剂中其他辅料对测定不干扰。取供试品在150 ℃干热条件下进行降解破坏,其降解后色谱图见图2,降解峰与供试品峰的分离度达2.7,可有效分离,不影响供试品测定。
图1 供试品的色谱图
1 辅料 2 克拉霉素
图2 150 ℃干热降解色谱图
1 辅料 2、3 降解峰 4 克拉霉素
3.2 由于克拉霉素不溶于水,其干混悬剂中又含有大量的辅料,故采用乙腈溶解后,加入磷酸盐缓冲液及甲醇,进行超声溶解,以此配制成的溶液,经实验,室温放置8 h,含量仍稳定。
, 百拇医药
3.3 大环内酯类抗生素本身紫外吸收不明显,HPLC法多采用末端吸收波长210 nm,由于甲醇吸收大,若移动相中采用40%的甲醇,基线测定不稳,重现性差。经实验,移动相按V(0.067 mol/L磷酸二氢钾):V(乙腈):V(甲醇)=63∶32∶5的配比,既能得到稳定的基线,又达到有效的分离,结果满意。
参考文献
[1]Gan VN,Chu SY,Kuamiesz HT, et al. Pharmacokinetics of a clarithromycin suspension in Infants and children[J]. AAC,1992,36(11):2478
[2]USP 24[S].425.
(收稿日期 2000-05-22), 百拇医药