米安色林药理学机制及临床应用研究进展
氟西汀,阿米,抗抑郁,1药理学机制,2临床研究
郭放 姚远 易正辉米安色林于1966年合成,刚开始的资料并没有显示其可能的抗抑郁作用,但随后的临床药理学研究表明它是一种有效的抗抑郁药,并且与三环类抗抑郁药相比,有更少的抗胆碱能作用和心脏毒性[1],米安色林和米氮平一样,同属NE能及特异性5-HT受体拮抗剂(NaSSA)类抗抑郁药物,通过阻断去甲肾上腺素α2自受体和异受体,选择性阻断5-HT2型受体和5-HT3型受体,同时阻断H1受体来发挥其抗抑郁、抗焦虑、促睡眠的作用,不过与米氮平不同,米安色林还具有强效的α1受体拮抗作用,α1受体拮抗作用联合H1受体拮抗作用,有效阻断脑内唤醒系统,镇静作用更为明显[2]。国外有关米氮平的临床研究较多,而米安色林研究较少,国内近年来有关米安色林的研究日益增多,因此本文对米安色林的药理作用机制及临床研究进行综述。
1 药理学机制

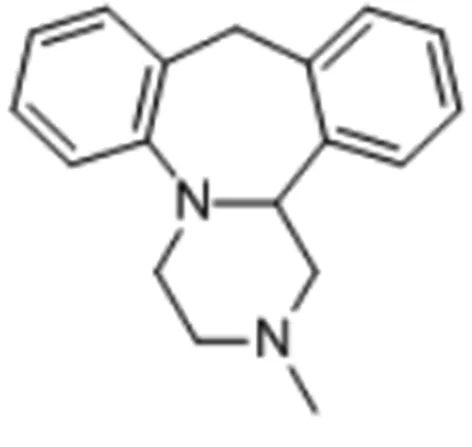
图1 米安色林化学结构
盐酸米安色林的分子量为300.08。米安色林为NE能及特异性5-HT受体拮抗剂(NaSSA),能阻断去甲肾上腺素α2自受体和异受体,增加NE和5-HT在突触间隙的浓度,起到抗抑郁、抗焦虑、促进睡眠作用,并能选择性阻断5-HT2型受体和5-HT3型受体,减少性功能障碍和恶心呕吐的副作用,同时阻断H1受体,具有镇静作用。但其并不能抑制去甲肾上腺素的再摄取。米安色林有左旋和右旋两种对映体,其都有药理活性,但两种对映体的抗抑郁活性都比消旋体低,因而临床使用的是消旋体。米安色林口服吸收后2 h在体内达到峰值(Cmax),主要分布于脑、心、肝等器官,血浆的半衰期为(9.6±2.1)h,清除半衰期为(16.1±6.8)h[3]。米安色林生物利用度较低,约为30%,有首过效应。血浆蛋白结合率96%,口服6 d达稳态血浓度,治疗血浓度为25~70 ng/ml[4]。95%的米安色林经肝脏代谢,途径为去甲基、8-羟化和N-氧化,最后由肾脏排出。其中,去甲基代谢物对突触前α受体的拮抗比米安色林稍弱;而对5-HT再摄取的抑制比米安色林稍强,但比丙咪嗪弱得多。8-羟化代谢物对5-HT和NE的摄取均无抑制作用 ......
您现在查看是摘要页,全文长 13949 字符。