欧盟GMP附录15确认与验证解析
生命周期,草案,评估,1新版附录15草案与2001版对比分析,1组织与计划,2文件编制,3设备,厂房设施和公用工程确认阶段,4工艺验证,5清洁验证,6其它方面,2中国意见稿与新版附录15草案对比浅析,1验证计划,3确认,3结束
苏丽花 郑金旺(上海东富龙科技股份有限公司,上海 201108)
2014 年2 月6 日欧盟委员会发布了欧盟药品生产管理规范(EU GMP)附录15-确认与验证草案[1](以下简称新版附录15 草案),该版本与2001 年9 月颁布的确认与验证[2]初始版本(以下简称老版附录15)有着显著的不同。该草案公开征求意见已于5 月结束,收到包括澳大利亚质量授权委员会AQPA、国际制药工程协会ISPE 等权威机构的诸多建议。与此同时,中国食品药品监督管理总局于2014 年6 月17 日再次发布2010 版GMP 附录确认与验证征求意见稿[3](以下简称中国意见稿),其引用新版附录15 草案相关内容和理念,但整体看来仍与老版附录15 接近。新版附录15 草案相对于老版本的主要变化有:
(1)参考EU GMP 修订版第Ⅰ部分“人用和兽用药品良好生产管理规范”[4]、EU GMP 附录11“计算机化系统”[5],引入国际人用药品注册技术要求协调会议ICH 已正式颁布的制药开发(Q8)[6]、质量风险管理(Q9)[7]、制药质量体系(Q10)[8]、原料药开发与生产(Q11)[9]。
(2)在设备/系统确认流程中,首次将用户需求说明URS、工厂验收测试FAT、现场验收测试SAT 纳入其中,作为确认的一部分。
(3)工艺验证参考欧盟药品管理局EMA 工艺验证指南[10,13],采用持续的、生命周期方法;清洁验证可接受标准基于毒理学评价确定产品特定的日允许暴露量PDE[11]。
(4)增加运输验证、包装验证、分析方法验证相关内容。
本文就新版附录15 草案与老版附录15 以及中国意见稿之间的不同要求进行简要分析。
1 新版附录15 草案与2001 版对比分析
新版附录15 草案从原来的11 页增加到17 页,目录对比如表1 所示:
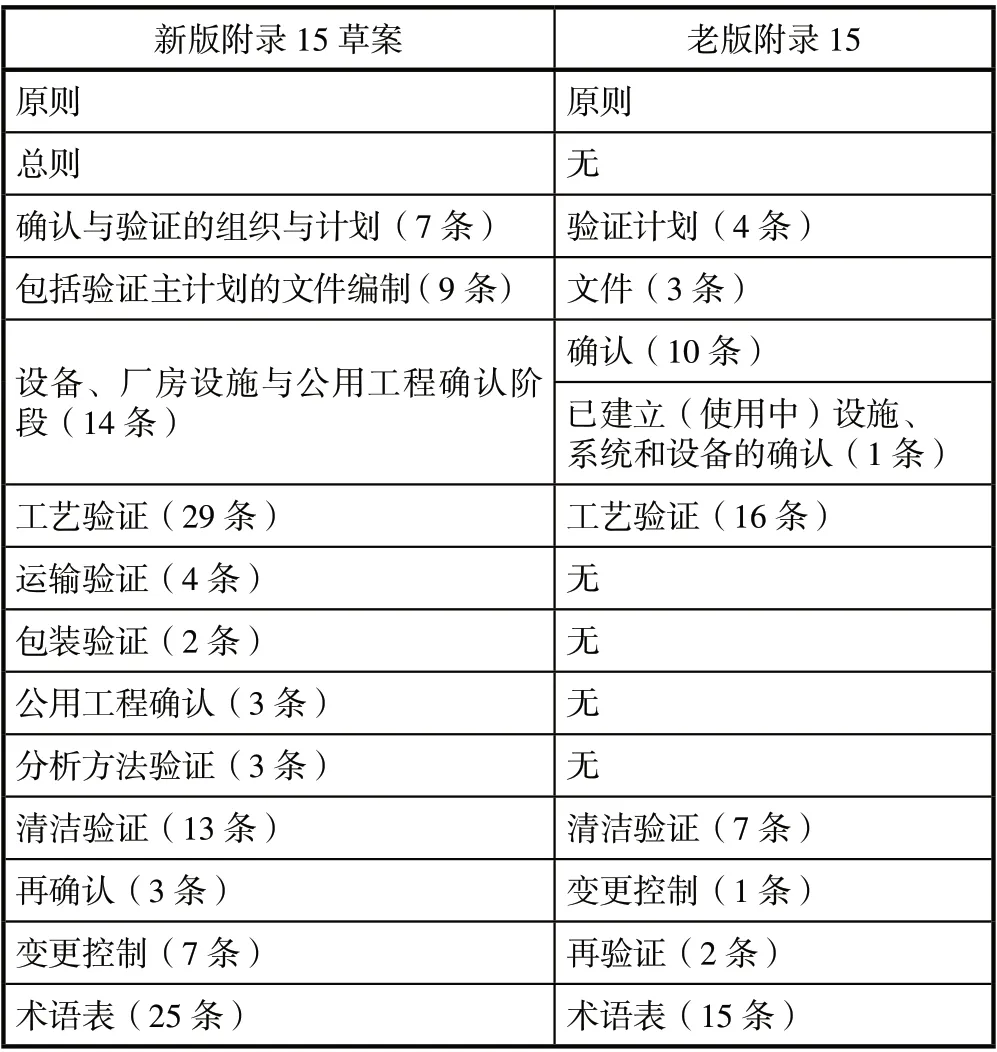
表1 新版附录15 草案与老版附录15 目录对比表
新版附录15 草案解决了确认与验证活动和产品/工艺相结合的问题,要求将质量风险管理方式应用于药品的整个生命周期,并通过风险评估方法确定验证与确认的范围和程度。用于药品生产的计算机化系统按照EU GMP 附录11 进行验证,并考虑ICH Q8、Q9、Q10、Q11 的相关概念和指导 ......
您现在查看是摘要页,全文长 19124 字符。