蛋白C p.Ala333Thr突变引起遗传性蛋白C缺陷症的分子机制
细胞器,突变体,质粒,1材料和方法,2结果,3讨论
余晓敏,常国林,郑逸旸,陈诚,唐施艺,吴鑫媛,吕佳,林向阳,朱丽青1.温州医科大学附属第一医院 医学检验中心 浙江省检验诊断及转化研究重点实验室,浙江 温州 325015;2.温州医科大学附属第一医院 温州市血液学重点实验室,浙江 温州 325015;3.温州医科大学附属第二医院 病理科,浙江 温州 325027
遗传性蛋白C缺陷症(inherited protein C deficiency, IPCD)是由位于2号染色体2q13-2q14的蛋白C基因(protein C gene, PROC)突变所引起的常染色体隐性、显性或不完全显性遗传性疾病[1],全球发病率约为1/16 000[2]。IPCD的典型临床症状包括血栓性并发症、暴发性紫癜、弥漫性血管内凝血和新生儿期继发性出血,与PROC突变类型密切相 关[3]。截至目前,已报道300多种PROC突变[4]。IPCD可分为两种类型:I型表现为蛋白C(protein C, PC)抗原水平和功能同时降低,II型表现为PC功能降低而抗原水平正常,或PC活性远低于抗原水平,大多数IPCD属于I型缺陷[5]。虽然IPCD于1981年就曾被首次报道[6],但其具体致病机制至今尚未完全明晰。本课题组发现了1个由p.Ala333Thr突变引起的I型IPCD家系,家系成员中多位有血栓形成史[7],经测序发现患者PROC的第9号外显子存在c.1084G>A杂合错义突变,导致p.Ala333Thr,但基因突变导致PC含量降低的分子机制尚不明确。因此,本研究构建PC p.Ala333Thr突变型质粒,通过体外实验对PC p.Ala333Thr突变导致IPCD的致病机制进行探讨。
1 材料和方法
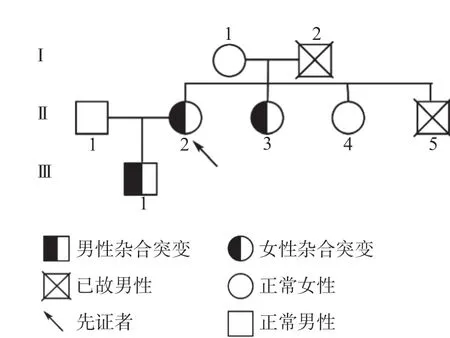
1.1 先证者及其家系 先证者及其家系6人均纳入研究,随机抽取本院100名健康成年人作为对照组,其中男45例,女55例,年龄12~58岁。本研究通过温州医科大学附属第一医院的伦理委员会审查(伦理批号:KY2022-R050),且所有受试者均知情并签署知情同意书。先证者,女,39岁,因双下肢反复疼痛前往温州医科大学附属第一医院就诊。既往有下肢深静脉血栓形成史,长期口服华法林抗凝,家族中父亲因脑静脉窦血栓死亡,哥哥因肺栓塞死亡,家系图见图1。入院检查:PC活性39%,抗核抗体1∶320阳性,下肢血管超声显示左小腿深静脉血栓形成,临床诊断为“PC缺陷症,系统性红斑狼疮,下肢深静脉血栓形成”。
 ......
......
您现在查看是摘要页,全文长 9773 字符。